この記事はPerl Advent Calendar 2017の14日目の記事です。
先月始めにアドベントカレンダーを知ったのですが、どの記事も面白くてためになりますね!
その流れをぶった切って、Bio::Perlの苦い経験を書いてみました。
序盤の文章は、ほぼPerlと関係ありません。
ニッチな分野でにっちもさっちも行かなかった、消化不良な結果であることをご了承ください。
解決策がもらえると嬉しいです!
始めに
現在、バイオインフォマティクス1という名のもとに、Ensembl2から取得したラットの塩基配列3のうち、snoRNA4群をNCBI5のサイト6でブラスト7しています。
今回は、ブラスト対象、被ブラスト対象がどちらもラットの塩基配列8でなければなりません(重要)。
この全自動化を試してみました。
理想郷

PlantUML文

以下、各アクションについて説明します。
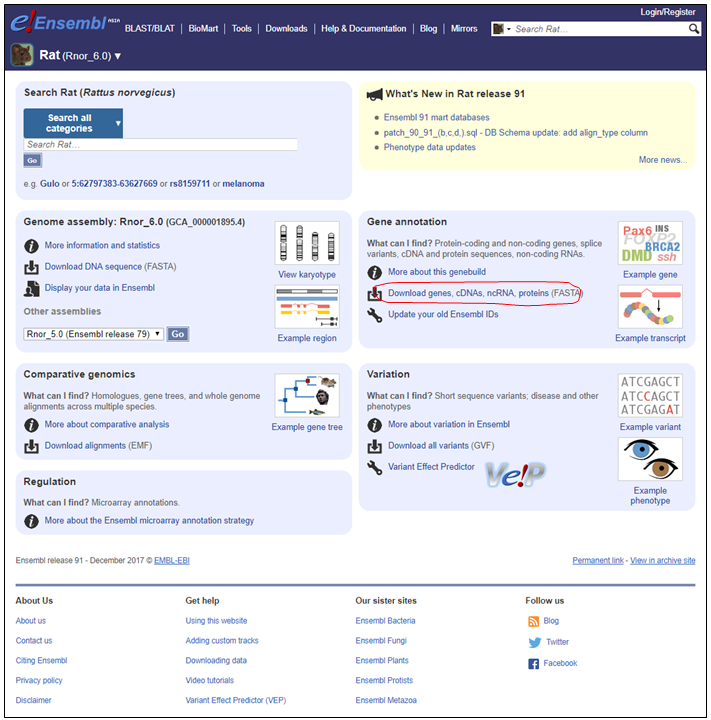
1. Ensemblのサイトからラットのゲノム情報を取得する
Ensemblでは、生体情報をFASTA形式9で公開しています。
https://www.ensembl.org/Rattus_norvegicus/Info/Index にあるDownload genes, cDNAs, ncRNA, proteinsリンク先のncrnaディレクトリ内に置いてある、Rattus_norvegicus.Rnor_6.0.ncrna.fa.gzをダウンロードし、解凍します10。
最初のページ以降はftp接続です。
下図に、最初のページを示します。
2. ラットのゲノム情報のうちsnoRNAに関するゲノム情報を抽出する
先ほど1. で取得したFASTA形式ファイルのコメント欄に、snoRNAと記述されたゲノム配列11を逐次取得します。
例えば、次のようなデータがある場合、2つ目のENSRNOT00000082727.1のゲノム配列のみを抽出します。
...
>ENSRNOT00000082553.1 ncrna chromosome:Rnor_6.0:9:92936066:92936231:1 gene:ENSRNOG00000053524.1 gene_biotype:snRNA transcript_biotype:snRNA gene_symbol:U1 description:U1 spliceosomal RNA [Source:RFAM;Acc:RF00003]
ATACTTACCTGGCAGGGGAGACACCATGATCATGGAAAGTGGTTTTCCTAGGGTGAGGCT
TATCCATTGAACTCCTGATGTGCTGACCCCTGTGATTTCCCCAAATGCTGGGAAACTCGA
CTGCATAATTTGTGATAATGGGGGACTGTGTTTGTGCTCTCCCCTG
>ENSRNOT00000082727.1 ncrna chromosome:Rnor_6.0:2:74322795:74322926:1 gene:ENSRNOG00000056734.1 gene_biotype:snoRNA transcript_biotype:snoRNA gene_symbol:SNORA17 description:Small nucleolar RNA SNORA17 [Source:RFAM;Acc:RF00560]
CCTGGCCTTTCAGGCACTGCAGATGTCGCTGCTGTGTCATATGTGTGTCAATAGGTGGCA
GAGATGAGAGACGCTCTGACTATGCTCAGTGTTCTGACCTAGGAACATCCGAATGATCAA
TGGCCTAACATT
...
ちなみに、2017/12/14現在に取得したRattus_norvegicus.Rnor_6.0.ncrna.faには、80000行を超えるゲノム配列が存在します。
そのうち、snoRNAは1654個含まれています。
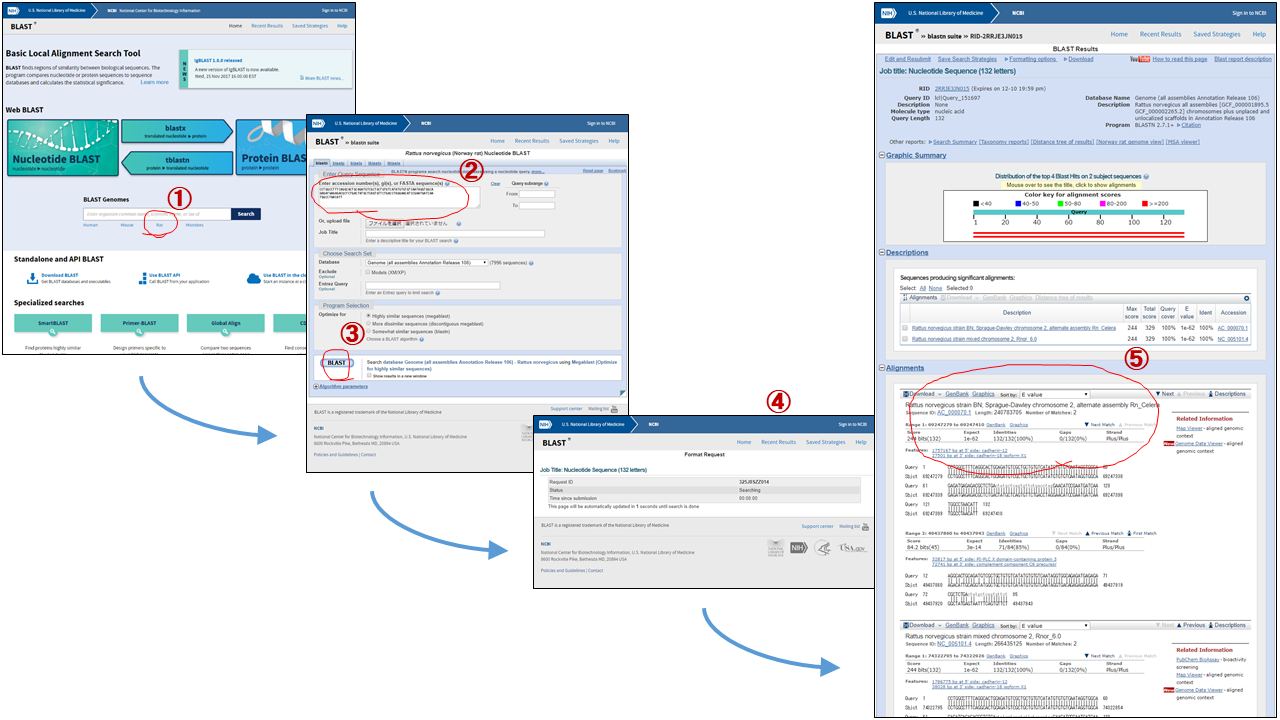
3. snoRNAゲノムをブラストする
NCBIのサイトで、ゲノム配列をブラストします12。
- NCBIのBLASTページから、ラットのデータベースからのみブラストするサイトに移動します(上辺に Rattus norvegicus (Norway rat) Nucleotide BLAST と書かれていることを確認してください)。
- ブラストしたいゲノム配列を
Enter accession number(s), gi(s), or FASTA sequence(s)の窓に貼り付けます。 - BLASTボタンを押します。
- しばらくお待ちください13。
- 検索結果が出てきたら、その中で検索した配列と完全一致したゲノム14の情報を取得します。
簡単な画面遷移図は、以下の通りです。
4. ブラスト結果をCSVファイルにまとめる
ブラスト結果のうち、シーケンスIDとゲノムの位置(始点と終点と向き)をCSV形式で出力します。
例えば、3.5.のページの、赤丸で囲まれた箇所を拡大してみます。
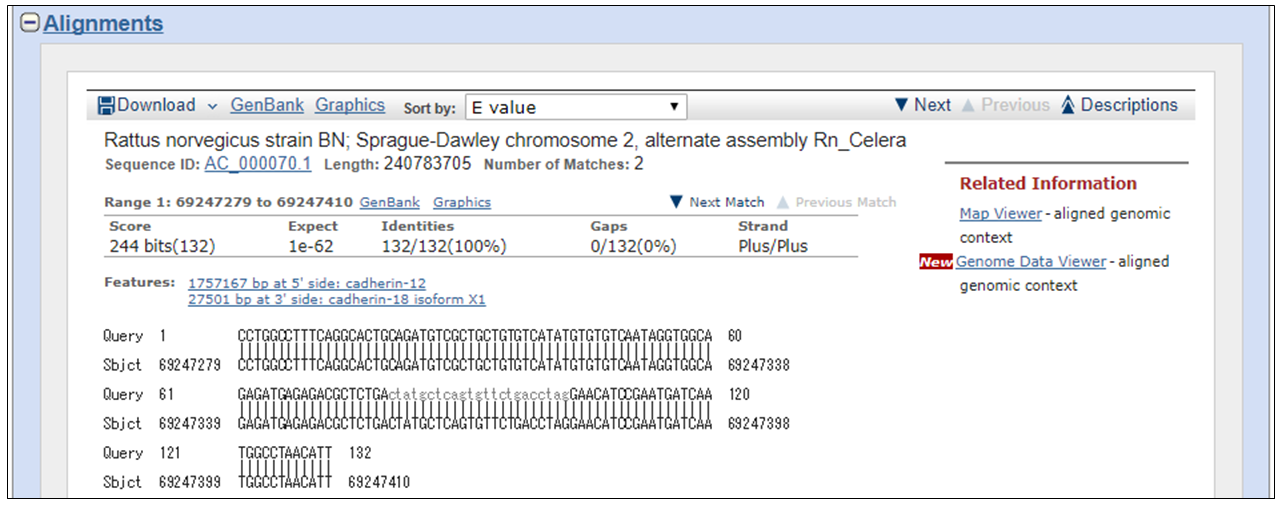
ここから、次のようなデータを抽出します。
なお、最初の行はカラム名を入れています。
PRIMARY_ACC,PRIMSPAN5P,PRIMSPAN3P,COMP
AC_000070.1,69247279,69247410,Plus
これを、Ensemblから取得した全snoRNAゲノムをブラストし、そのブラスト結果のデータを全部入れたCSVファイルを生成します。
ここから、ようやくPerlの話に入ります。
実は、シンプルなブラスト実行であれば、Bio::Perl15を利用できます。
例えば、先ほどのゲノム配列をブラストするならば、次のようなコードだけで簡単にデータを抽出できます。
use strict;
use warnings;
use Bio::Perl;
use Data::Dumper; # データ可視化用
my $sequence = "CCTGGCCTTTCAGGCACTGCAGATGTCGCTGCTGTGTCATATGTGTGTCAATAGGTGGCAGAGATGAGAGACGCTCTGACTATGCTCAGTGTTCTGACCTAGGAACATCCGAATGATCAATGGCCTAACATT";
my $blast_result = Bio::Perl->blast_sequence($sequence);
print Dumper $blast_result;などを行えば、$blast_resultが入り組んだハッシュであることがわかると思います16。
例えば、次のようなデータ17には...
print Dumper $blast_result->{_iterations}->[0]->{_newhits_below_threshold}->[0];
$VAR1 = {
'-rank' => 1,
'-accession' => 'LN871987.1',
'-hsp_factory' => bless( {
'_root_verbose' => 0,
'interface' => 'Bio::Search::HSP::HSPI',
'type' => 'Bio::Search::HSP::GenericHSP',
'_loaded_types' => {
'Bio::Search::HSP::GenericHSP' => 1
}
}, 'Bio::Factory::ObjectFactory' ),
'-description' => 'TPA: Mus musculus SNORA17 (ACA17) gene for small nucleolar RNA SNORA17 (ACA17)',
'-hsps' => [
{
'-query_end' => '132',
'-conserved' => '111',
'-query_seq' => 'CTGGCCTTTCAGGCACTGCAGATGTCGCTGCTGTGTCATATGTGTGTCAATAGGTGGCAGAGATGAGAGACGCTCTGACTATGCTCAGTGTTCTGACCTAGGAACATCCGAATGATCAATGGCCTAACATT',
'-query_length' => '132',
'-hit_frame' => 0,
'-algorithm' => 'BLASTN',
'-hit_length' => '132',
'-hsp_gaps' => '0',
'-query_start' => '2',
'-hit_name' => 'LN871987.1',
'-homology_seq' => '||| || | ||||| ||||| ||| |||||||||||| |||||||||| ||||||||||||| |||||| ||| || ||| ||||||||||||||||| ||||||| ||||||| | | ||||||||||',
'-bits' => '147',
'-hsp_length' => '131',
'-rank' => 1,
'-query_frame' => 0,
'-evalue' => '7e-32',
'-hit_seq' => 'CTGCCCCTACAGGCGTTGCAGGTGTGGCTGCTGTGTCACATGTGTGTCAGTAGGTGGCAGAGAGGAGAGAGGCTATGTCTACGCTCAGTGTTCTGACCTGTGAACATCTGAATGATTAGTAGCCTAACATT',
'-query_desc' => '',
'-score' => '162',
'-hit_start' => '2',
'-hit_end' => '132',
'-identical' => '111',
'-hit_desc' => 'TPA: Mus musculus SNORA17 (ACA17) gene for small nucleolar RNA SNORA17 (ACA17)',
'-query_name' => 'blast-sequence-temp-id'
}
],
'-algorithm' => 'BLASTN',
'-query_len' => '132',
'-length' => '132',
'-significance' => '7e-32',
'-name' => 'LN871987.1'
};
という出力結果を得ることができます。
であれば、次のようにすればいいのではないでしょうか?
my $sequence_id = $blast_result->{_iterations}->[0]->{_newhits_below_threshold}->[0]->{'-accession'};
my $start_position = $blast_result->{_iterations}->[0]->{_newhits_below_threshold}->[0]->{'-hsps'}->[0]->{'-hit_start'};
my $end_position = $blast_result->{_iterations}->[0]->{_newhits_below_threshold}->[0]->{'-hsps'}->[0]->{'-hit_end'};
my $strand = 'Plus';
if ($end_position < $start_position) {
($start_position, $end_position) = ($end_position, $start_position);
$strand = 'Minus';
}
意外と簡単にブラストできました!
...本当でしょうか?
現実
理想は現実になりませんでした。
結論を先に言うと、3.のsnoRNAゲノムのブラストが上手くいきませんでした。
問題点
説明文を見てください。
print $blast_result->{_iterations}->[0]->{_newhits_below_threshold}->[0]->{'-hsps'}->[0]->{'-hit_desc'}, "\n";
TPA: Mus musculus SNORA17 (ACA17) gene for small nucleolar RNA SNORA17 (ACA17)
なんとこれ、ラットのゲノムのブラスト結果ではなく、マウスのゲノムのブラスト結果だったみたいです18!
最初に、ラットの塩基配列をラットのゲノム情報からブラストする、という旨を書きました。
しかしこの場合は、ラットの塩基配列を__マウス__のゲノム情報からブラストしてしまっています。
これでは正しい情報が取得できません。
では、何が問題だったのでしょうか?
ブラスト先のURLが異なる
いろいろ調べてみた結果、どうやらBio::Perlではスタンダードなブラスト(一般的な動物のゲノム情報を沢山詰め込んだデータベースからブラストしているっぽい)しかできないようです。
スタンダードなブラストを行うサイトと、ラットのゲノム情報からブラストするサイトのURLを見ると、クエリが所々異なります。
standard > https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome
rattus > https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch&BLAST_SPEC=OGP__10116__10621&LINK_LOC=blasthome
ブラスト対象のデータベースが異なる
これは、2つのサイトをいろいろ見比べてみた結果なのですが、BLAST実行ボタンの横に書かれている検索先が異なります。
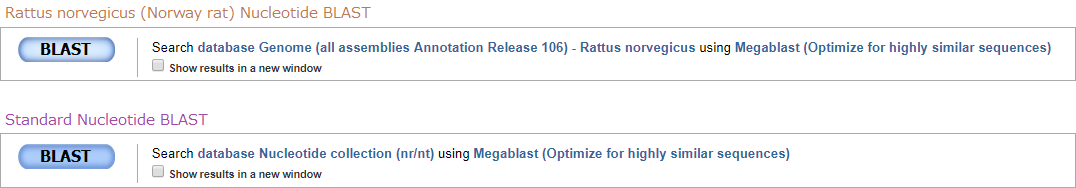
試行錯誤
ここまでで僕の心は半分折れていましたが、いろいろ頑張ってみました。
ブラスト先のURL問題
URLが異なるのであれば、異なるURL先を見るように変更すればいいのです。
Bio::Perl内部を調べた結果、Bio::Tools::Run::RemoteBlastが問題なのではないかと考えました。
# ...
sub blast_sequence {
my ($seq,$verbose) = @_;
# ...
require Bio::Tools::Run::RemoteBlast;
# ...
my $factory = Bio::Tools::Run::RemoteBlast->new(@params);
# ...
LOOP :
while( my @rids = $factory->each_rid) {
foreach my $rid ( @rids ) {
my $rc = $factory->retrieve_blast($rid); # 犯人はお前だ
if( !ref($rc) ) {
if( $rc < 0 ) {
$factory->remove_rid($rid);
}
if( $verbose ) {
print STDERR ".";
}
sleep 10;
} else {
$result = $rc->next_result();
$factory->remove_rid($rid);
last LOOP;
}
}
}
# ...
return $result;
}
# ...
更に、そのBio::Tools::Run::RemoteBlast内では、URLの指定がありました。
# ...
sub retrieve_blast {
my($self, $rid) = @_;
my $url_base = $self->get_url_base;
my %hdr = %RETRIEVALHEADER;
$hdr{'RID'} = $rid;
my $req = HTTP::Request->new(
GET => $url_base."?CMD=Get&FORMAT_OBJECT=SearchInfo&RID=$rid",
);
# ...
}
# ...
また、$url_base eq "https://blast.ncbi.nlm.nih.gov/Blast.cgi"であることも突き止めました。
ということは、Bio::Tools::Run::RemoteBlastのURLを設定している箇所だけ変更すればよいだけです。
しかし、cpanmでダウンロードしたモジュールを上書きするという方法を、当時の僕は知りませんでした。
ということで、Bio::Tools::Run::RemoteBlastを継承したRemoteBlastByRat.pmを作り、それを用いるためにBio::Perlを継承したBioPerlByRat.pmを作りました。
関数内はほぼコピペです。
変更箇所は、RemoteBlastByRat.pmでは「試行錯誤箇所」と「追加箇所」、BioPerlByRat.pmでは引数とrequire文とnew文です。
構文がわかりずらいですが、変更するのも怖いのでそのままにしました。
package RemoteBlastByRat;
use HTTP::Request::Common; # これがないとPOSTが動かなかった
use base 'Bio::Tools::Run::RemoteBlast';
# これがないと%hdrが空っぽだった
our %RETRIEVALHEADER = (
'CMD' => 'Get',
'ALIGNMENTS' => '1',
'ALIGNMENT_VIEW' => 'Pairwise',
'DESCRIPTIONS' => '5',
'FORMAT_TYPE' => 'Text',
'DATABASE' => 'Genome (all assemblies Annotation Release 106) - Rattus norvegicus',
'BLAST_SPEC' => 'OGP__10116__10621',
'LINK_LOC' => 'blasthome',
);
sub retrieve_blast {
my($self, $rid) = @_;
my $url_base = $self->get_url_base;
my %hdr = %RETRIEVALHEADER;
$hdr{'RID'} = $rid;
# 試行錯誤箇所-->
my $req = HTTP::Request->new(
# GET => $url_base."?CMD=Get&FORMAT_OBJECT=SearchInfo&RID=$rid", # 元URL
# GET => $url_base."?PAGE_TYPE=BlastSearch&BLAST_SPEC=OGP__10116__10621&LINK_LOC=blasthome&CMD=Get&FORMAT_OBJECT=SearchInfo&RID=$rid", # 変更URLその1
GET => $url_base."?CMD=$hdr{ 'CMD' }&RID=$hdr{ 'rid' }", # 変更URLその2
);
# これも追加しないと、何も出力されない
$req->{ '_content' } = "CMD=$hdr{ 'CMD' }&RID=$hdr{ 'rid' }&DESCRIPTIONS=$hdr{ 'DESCRIPTIONS' }&FORMAT_TYPE=$hdr{ 'FORMAT_TYPE' }&ALIGNMENTS=$hdr{ 'ALIGNMENTS' }&ALIGNMENT_VIEW=Pairwise";
$req->{ '_headers' }->{ 'content-length' } = length $req->{ '_content' };
# ここまで-->
# $self->debug("SearchInfo request is " . $req->as_string());
my $response = $self->ua->request($req);
if( $response->is_success ) {
my $status;
if($response->content =~ /Status=(WAITING|ERROR|FAILED|UNKNOWN|READY)/i ) {
$status = $1;
if( $status eq 'ERROR' ) {
$self->warn("Server Error");
return ERR_QBSTATUS;
} elsif( $status eq 'FAILED' ) {
$self->warn("Request Failed");
return ERR_QBSTATUS;
}
} else {
$self->warn("Error: No status reported\n");
}
if ( $status ne 'READY' ) {
return 0;
} else {
my ($fh,$tempfile) = $self->tempfile();
close $fh;
my $req = POST $url_base, [%hdr];
$self->debug("retrieve request is " . $req->as_string());
# 追加箇所-->
$req->{ '_content' } = "CMD=$hdr{ 'CMD' }&RID=$hdr{ 'rid' }&DESCRIPTIONS=$hdr{ 'DESCRIPTIONS' }&FORMAT_TYPE=$hdr{ 'FORMAT_TYPE' }&ALIGNMENTS=$hdr{ 'ALIGNMENTS' }&ALIGNMENT_VIEW=Pairwise";
$req->{ '_headers' }->{ 'content-length' } = length $req->{ '_content' };
# ここまで-->
my $response = $self->ua->request($req, $tempfile);
my $blastobj;
my $mthd = $self->readmethod;
$mthd = ($mthd =~ /blasttable/i) ? 'blasttable' :
($mthd =~ /xml/i) ? 'blastxml' :
($mthd =~ /pull/i) ? 'blast_pull' :
'blast';
$blastobj = Bio::SearchIO->new(
-file => $tempfile,
-format => $mthd);
## store filename in object ##
$self->file($tempfile);
return $blastobj;
}
} else {
$self->warn($response->error_as_HTML);
return ERR_HTTPFAIL;
}
}
1;
package BioPerlByRat;
use base 'Bio::Perl';
use RemoteBlastByRat;
sub blast_sequence {
# my ($seq,$verbose) = @_;
my ($self,$seq,$verbose) = @_; # 変更
if( !defined $verbose ) {
$verbose = 1;
}
if( !ref $seq ) {
$seq = Bio::Seq->new( -seq => $seq, -id => 'blast-sequence-temp-id');
} elsif ( !$seq->isa('Bio::PrimarySeqI') ) {
croak("[$seq] is an object, but not a Bio::Seq object, cannot be blasted");
}
# require Bio::Tools::Run::RemoteBlast;
require RemoteBlastOriginalUrl; # 変更
my $prog = ( $seq->alphabet eq 'protein' ) ? 'blastp' : 'blastn';
my $e_val= '1e-10';
my @params = (
'-prog' => $prog,
'-expect' => $e_val,
'-readmethod' => 'SearchIO' );
# my $factory = Bio::Tools::Run::RemoteBlast->new(@params);
my $factory = RemoteBlastOriginalUrl->new(@params); # 変更
my $r = $factory->submit_blast($seq);
if( $verbose ) {
print STDERR "Submitted Blast for [".$seq->id."] ";
}
sleep 5;
my $result;
LOOP :
while( my @rids = $factory->each_rid) {
foreach my $rid ( @rids ) {
my $rc = $factory->retrieve_blast($rid);
if( !ref($rc) ) {
if( $rc < 0 ) {
$factory->remove_rid($rid);
}
if( $verbose ) {
print STDERR "$count ";
}
sleep 10;
} else {
$result = $rc->next_result();
$factory->remove_rid($rid);
last LOOP;
}
}
}
if( $verbose ) {
print STDERR "\n";
}
return $result;
}
1;
...ですが、全く変化がありませんでした。
いろいろ調べてみたのですが、どういうわけかURLはブラウザ版とCUIツール版でそもそも異なるようです。
ということは、ラットのゲノム情報からブラストするサイトのURLに書き換えたところで、それはブラウザ版のURLであるためにそもそも見てくれません。
ここで僕の心はほぼ折れました。
ブラスト先のデータベース問題
データベースが異なるのであれば、異なるデータベースを検索するように変更すればいいのです。
ほぼ折れた心で再びBio::Tools::Run::RemoteBlastを見ると、データベースの指定ができそうです。
# ...
=head2 database
Title : database
Usage : my $db = $self->database
Function: Get/Set the database to search. Retained for backwards-compatibility.
Returns : string
Args : string [ swissprot, nr, nt, etc... ]
=cut
sub database {
my ($self, $val) = @_;
return $self->submit_parameter('DATABASE',$val);
}
# ...
しかもこれはクラス属性っぽいですから、Bio::Tools::Run::RemoteBlastを継承したモジュールは必要なさそうです。
希望の光が見えた気がしたので、Bio::Perlを継承した別モジュールを作ってみました。
package BioPerlByRatChangedDatabase;
use base 'Bio::Perl';
sub blast_sequence {
# ...
my $factory = Bio::Tools::Run::RemoteBlast->new(@params);
# 追加箇所-->
# ここ以外は元コードと同じ(先ほどのBioPerlByRat.pmと似たような書き方)
$factory->database("Genome (all assemblies Annotation Release 106)"); # この引数はいろいろ変えてみました
# ここまで-->
my $r = $factory->submit_blast($seq);
#...
}
1;
...こちらも思うようにはいきませんでした。
PODの引数の説明欄をよく見ると、次のように書いてあります。
Args : string [ swissprot, nr, nt, etc... ]
これ、NCBIの検索先データベースではなく、検索するサイト自体のことみたいです。
実は、ブラストしたりするための生物学的データベースは、NCBIに限らず様々あります。
つまり、ここで設定するのは大きな意味でのデータベースであり、NCBI内での検索先データベースとは全く違うものでした。
その内部のデータベース(あるいは別の名前があるかもしれません)を設定できる箇所が見つかればいいのですが、僕の力では見つけられませんでした。
僕の心はぽっきり折れてしまいました。
現状
全自動snoRNA情報ブラスト機の夢は、もろくも崩れ去ることとなりました。
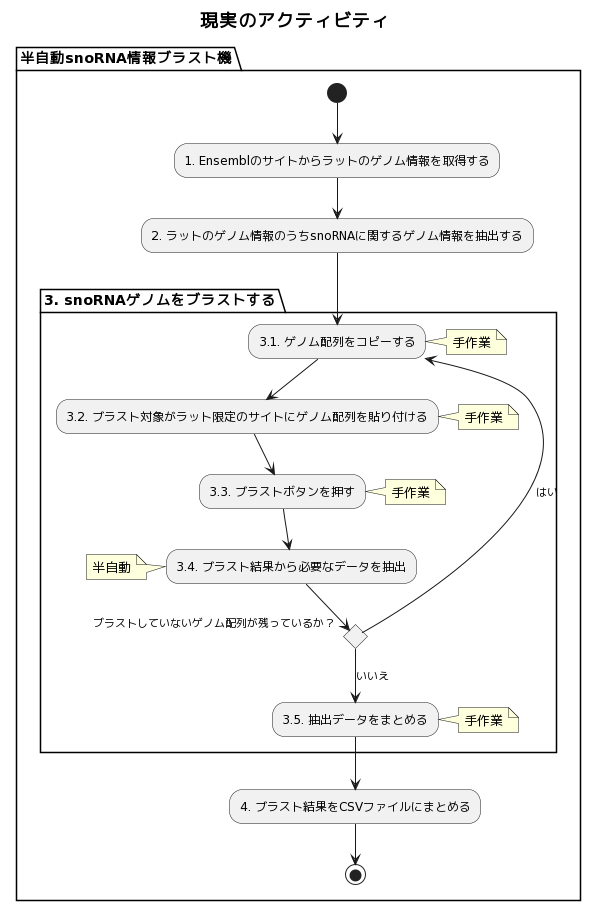
PlantUML文

それでも、一部抵抗が見られます。
3.4. ブラスト結果から必要なデータを抽出
ブラスト結果のページより、適当に情報をコピーします。
Sprague-Dawley chromosome 2, alternate assembly Rn_Celera
Sequence ID: AC_000070.1Length: 240783705Number of Matches: 2
Related Information
Map Viewer-aligned genomic context
NewGenome Data Viewer-aligned genomic context
Range 1: 69247279 to 69247410GenBankGraphicsNext MatchPrevious Match
Alignment statistics for match #1
Score Expect Identities Gaps Strand
244 bits(132) 1e-62 132/132(100%) 0/132(0%) Plus/Plus
Features:
1757167 bp at 5' side: cadherin-12
27501 bp at 3' side: cadherin-18 isoform X1
ここから、必要な情報4つを抽出するCUIシステムを作りました。
このシステムは、引数にsnoRNAゲノム情報を抽出する回数19を入力して実行し、上記のプレーンテキストをコピペすると、必要な情報4つを抽出します。
その回数情報を抽出した後、改行を5回連続で入力すると、1つのCSVファイルとして出力します。
まだMouseモジュール20などを知らなかったころのコードなので、読みずらいと思います21。
use strict;
use warnings;
use BlastResultParser;
my $parse_count = $ARGV[ 0 ];
print STDERR "Please input parsing lines $parse_count time(s).\n\n";
my $hash_list_ref = extract_blast_result_list( $parse_count );
my $writing_list_ref = format_writing_text_list_from_hash_list( $hash_list_ref );
write_text_from_list( $writing_list_ref );
exit(0);
# ブラスト結果を抽出します
sub extract_blast_result_list {
my $parse_count = shift;
my @blast_results;
for( my $i = 0; $i < $parse_count; $i++ ) {
my $result = parse_blast_result();
push @blast_results, $result;
print STDERR "\n\n[".$i+1."] >> Get [Sequence ID:$result->{ sequence_id }]'s data.\n\n\n";
}
ignore_unnecessary_input_line();
return \@blast_results;
}
# ブラスト結果のプレーンテキスト文から情報をパースします
sub parse_blast_result {
my $parser = BlastResultParser->new();
my @original_text_lines;
while( 1 ) {
chomp( my $line = <STDIN> );
next if not $parser->has_started_parse_line( $line );
# 解析ライン挿入開始
push @original_text_lines, $line;
# 解析ライン挿入終了
last if $parser->is_ending_parse_line( $line );
}
$parser->{ original_text_ref } = \@original_text_lines;
$parser->parse_original_text();
return $parser;
}
# データ抽出後に入力される不必要なコピペ文を無視します
sub ignore_unnecessary_input_line {
my $line_count = 0;
print STDERR "> OK, input data is finished!\n";
print STDERR "> Please input newline char 5 counts.\n\n\n";
# 5回連続空行の場合はプログラム終了
while( $line_count < 5 ) {
chomp( my $line = <STDIN> );
if( length( $line ) <= 0 ) {
$line_count++;
} else {
$line_count = 0;
}
}
}
# 出力ファイルを整形します
sub format_writing_text_list_from_hash_list {
my $ref = shift;
my @writing_list;
push @writing_list, "PRIMARY_ACC,PRIMSPAN5P,PRIMSPAN3P,COMP";
foreach my $hash ( @$ref ) {
push @writing_list, "$hash->{ sequence_id },$hash->{ from },$hash->{ to },$hash->{ strand }";
}
push @writing_list, "END";
return \@writing_list;
}
# ファイルを出力します
sub write_text_from_list {
my $ref = shift;
my $file_name = "blast_data.csv";
open( FILE, ">", $file_name ) or die "$!";
foreach my $list ( @$ref ) {
print FILE $list, "\n";
}
close FILE;
print STDERR "OK, Succeed to write file in $file_name\n";
}
package BlastResultParser;
use strict;
use warnings;
sub new {
my $class = shift;
my $self = {
is_parsing => 0,
original_text_ref => '',
sequence_id => '',
length => 0,
number_of_matches => 0,
range => 0,
from => 0,
to => 0,
strand => '',
};
return bless $self, $class;
}
sub has_started_parse_line {
my $self = shift;
my $line = shift;
if( not $self->{ is_parsing } and index( $line, "Sequence ID:" ) == 0 ) {
$self->{ is_parsing } = 1;
}
return $self->{ is_parsing };
}
sub is_ending_parse_line {
my $self = shift;
my $line = shift;
if( $self->{ is_parsing } and index( $line, "Plus/" ) > 0 ) {
$self->{ is_parsing } = 0;
}
return not $self->{ is_parsing };
}
# ブラスト結果のプレーンテキスト文から必要な情報を抽出します
sub parse_original_text {
my $self = shift;
foreach my $line ( @{ $self->{ original_text_ref } } ) {
if( index( $line, "Sequence ID:" ) == 0 ) {
$line =~ /Sequence ID: (.*)Length: ([0-9]*)Number of Matches: ([0-9]*)/;
$self->{ sequence_id } = $1;
$self->{ length } = $2;
$self->{ number_of_matches } = $3;
}
if( index( $line, "Range " ) >= 0 ) {
$line =~ /Range ([0-9]*): ([0-9]*) to ([0-9]*)/;
$self->{ range } = $1;
$self->{ from } = $2;
$self->{ to } = $3;
}
if( index( $line, "Plus/" ) > 0 ) {
$line =~ m|Plus/(.*)$|;
$self->{ strand } = $1;
}
}
}
1;
使用例を示します。
$ perl parsing_data.pl 2
Please input parsing lines 2 time(s).
適当にコピペしたプレーンテキスト文
Sprague-Dawley chromosome 2, alternate assembly Rn_Celera
Sequence ID: AC_000070.1Length: 240783705Number of Matches: 2
Related Information
Map Viewer-aligned genomic context
NewGenome Data Viewer-aligned genomic context
Range 1: 69247279 to 69247410GenBankGraphicsNext MatchPrevious Match
Alignment statistics for match #1
Score Expect Identities Gaps Strand
244 bits(132) 1e-62 132/132(100%) 0/132(0%) Plus/Plus
[1] >> Get [Sequence ID:AC_000070.1]'s data.
Features:
1757167 bp at 5' side: cadherin-12
27501 bp at 3' side: cadherin-18 isoform X1
ここまでコピペ文が入力される(ターミナル上でコピペすると、一連のデータを抽出した後もコピペ文が続く)
mixed chromosome 4, Rnor_6.0
Sequence ID: NC_005103.4Length: 184226339Number of Matches: 3
Related Information
Map Viewer-aligned genomic context
NewGenome Data Viewer-aligned genomic context
Range 1: 102646851 to 102646977GenBankGraphicsNext MatchPrevious Match
Alignment statistics for match #1
Score Expect Identities Gaps Strand
235 bits(127) 6e-60 127/127(100%) 0/127(0%) Plus/Plus
[2] >> Get [Sequence ID:NC_005103.4]'s data.
> OK, input data is finished!
> Please input newline char 5 counts.
Features:
2089 bp at 5' side: Ig kappa chain V-II region 26-10-like
18552 bp at 3' side: Ig kappa chain V-II region 2S1.3-like
Query 1 CCTGACTTATAGAGAATATGCAGATGTCAGTGCTGtatcacatgtgtgtttgtaagtgGC 60
||||||||||||||||||||
ここから5連続エンターキーを入力して終了する
OK, Succeed to write file in blast_data.csv
$ cat blast_data.csv
PRIMARY_ACC,PRIMSPAN5P,PRIMSPAN3P,COMP
AC_000070.1,69247279,69247410,Plus
NC_005103.4,102646851,102646977,Plus
END
考察
この挫折までに多くのことを勉強できました。
- Perlでの継承、オーバーライドを簡単ながら実装した
- デバッグモードを使ってモジュール内部まで見る方法を学んだ
- 複雑なハッシュや配列の組み合わせから必要なデータを抽出できた
- 他人が書いたソースコードを読んだ
- バイオインフォマティクスでの用語やツールを知った
- 英語マニュアルへの抵抗が減った
今思えば、この挫折も無駄ではなかったようです。
まとめ
バイオインフォマティクスはなかなか面白いです。
今でこそPythonにとって代わりつつありますが、未だにNCBIの提供するツールがPerlであったりと、この分野はまだPerlが強いと思います。
なにより僕は、Perlのコードを読めば読むほど、理解度が上がって楽しかったです。
ここまで読んでくださってありがとうございました!
もしおかしい点、気づいた点がありましたら教えてください。
特に、生物学全般やBio::Perlについてはあまり詳しくないため、誤解などが多々あると思います。
そして、僕のやりたいことが可能な別の手法があれば、ぜひ教えてください!!!
むしろこれが知りたくて記事を書いたといっても過言ではありません。
-
日本語に直訳すれば、生体情報科学です。つまり、情報科学の手法を駆使して生体情報を解析しよう、という学問です。 ↩
-
生物情報のデータベースです。サイトは https://www.ensembl.org/index.html です。 ↩
-
生物の遺伝子情報は4種類しかない塩基の組合せである、みたいな話を聞いたことはありませんか?DNAではAとTとCとGの4種類です。ややこしいことに生物学分野では、シーケンスとか文字列と言うこともあります。 ↩
-
ncRNAと呼ばれる、RNAの一種です。核小体に存在し、低分子であることが特徴です。詳しい説明は http://jhfsp.jsf.or.jp/frontier-science/newsletter/014/nl-04.html をご覧ください。 ↩
-
アメリカの国立生物工学情報センター (National Center for Biotechnology Information) です。 ↩
-
ホームページは https://www.ncbi.nlm.nih.gov/ ですが、今回利用するサイトはBLASTのページです。 ↩
-
BLAST (Basic Local Alignment Search Tool) とは、とある塩基配列あるいはアミノ酸配列の類似した領域を特定すること(シーケンスアラインメント)、またはそれを行うアルゴリズムやツールのことです。ブラストする、という動詞型でも通用します。 ↩
-
被ブラスト対象という言葉が正しいかどうかわかりませんが、簡単に説明すると、とあるラットの塩基配列をラットのゲノム情報からブラストする、という意味です。 ↩
-
最初の行に">"で始まるコメント、その次の行以降にゲノム配列を書いたプレーンテキストファイルです。ややこしいことに、FASTAと呼ばれるシーケンスアラインメントを行うソフトウェアも存在します。FASTAとBLASTの違いについて、詳しい説明は http://www.ddbj.nig.ac.jp/search/archives/homology_doc-j.html をご覧ください。 ↩
-
ディレクトリ名やその階層やファイル名は変わることがあります。 ↩
-
正確には、gene_biotypeオプションがsnoRNAであるゲノム配列(
gene_biotype:snoRNA)です。 ↩ -
詳しいチュートリアルは、 http://www.jst.go.jp/nbdc/bird/minicourses/blast-tutorial.pdf をご覧ください。 ↩
-
BLAST自体が時間のかかる手法である点、またNCBIのデータベースへネットワークを介している点から、長いときは数分かかります。更に、何度も検索すると数時間失敗し続けることもあります。ご利用は計画的に。 ↩
-
検索した塩基配列の長さとIdentitiesの長さが一致し、かつIdentitiesが100%であるゲノムのことを指します。 ↩
-
Bioperlプロジェクトについては公式サイト、BioPerlの簡単な使い方はCPAN、ソースコードはGitHubをご覧ください。 ↩
-
本来であれば、
write_blast(">output_file_name.txt", $blast_result);を利用することで、出力ファイル先にそれっぽく整形してくれるのですが、今回は整形してもらう必要がないので利用しません。 ↩ -
次のような配列のリファレンスのハッシュのリファレンスの配列のリファレンスのハッシュのリファレンスをデリファレンスしたデータ、ですね... ↩
-
ラットの学名はRattus norvegicus、マウスの学名はMus musculusです。4.のサイトの説明文にはRattus norvegicusと書いてあります。 ↩
-
snoRNAゲノムの個数と書かなかったのは、1回で数百個もの情報を抽出と結構な時間がかかるため、僕は複数回に分けて実行したからです。 ↩
-
当然、生物のマウスの話ではありません。 ↩
-
少しでも読みやすくするために、エラー時の対処に関する処理など、本質とは関係のない箇所は除きました。 ↩