はじめに
次世代シーケンサーが登場してはや15年強。少しネットを調べれば、染色体レベルでアセンブリされたシークエンスデータが手軽に手に入る時代になりました。
せっかく公開されているんです。使わなければ損! あなたのシーケンスデータと公開データを比較してみましょう。
しかし、シーケンスデータの処理はバイオが専攻、特にウェットの生物学が専攻の人には簡単ではないですね。
本記事では、TBtoolsというツールを使って、簡単にGUIでゲノムシンテニープロットを比較する方法を紹介します。
Genome assembly データと Gene annotation データがあればすぐできますので、この記事を見たらぜひやってみてください。あなたも染色体進化研究の虜になりますよ!
目次
TBtoolsとは
"Tools for Biologist"の略で、ハイスループットデータを扱うのに役立つツールキット。
さまざまな生物学的データ処理ツールを統合した生物学者向けのツールセットで、スタンドアロンのソフトウェアにより、GUIで使えることが特徴です。
具体的には、シーケンスの各種操作(ORF予測、変換など)、BLAST、GO・KEGG(アノテーション)、ベン図やヒートマップおよびシンテニープロットなどの図の作成などができます。
今回使用する、ゲノム間のシンテニー領域を調べる機能のアルゴリズムには、MCScanXを採用しています。
GitHub上にソースコードが公開されています。
https://github.com/CJ-Chen/TBtools
ベン図、ヒートマップ、シンテニーブロックの可視化など、分析結果の例がREADMEに載ってますので、参考にしてください。
コマンドラインからも実行できますが、今回は普段CUIに触らない生物学者向けですので、GUIベースでお話を進めます。
インストール
TBtools
GitHubの最新リリース からインストールしてください。
Windowsの場合は、ダウンロードした.exeファイルを実行して設定するだけです。
解析に使うシーケンスデータ
Ensembl Rapid Releaseから、適当な種の Genome assembly データ(FASTA) と Gene annotation データ(GFF3) を取ってきましょう。
(目的の種は一覧ページから探しましょう)
今回は例として、モンシロチョウ(cabbage white)とオオモンシロチョウ(large cabbage white)のデータを使いました。
| Name | Scientific name | Genome assembly | mask |
|---|---|---|---|
| モンシロチョウ | Pieris rapae | ilPieRapa1.1 (GCA_905147795.1) | hardmasked |
| オオモンシロチョウ | Pieris brassicae | ilPieBrab1.1 (GCA_905147105.1) | hardmasked |
ダウンロードされるのは圧縮ファイル(.gz)ですので、解凍をお忘れなく。
自分のデータを使う場合も、FASTAファイルと、GFF3もしくはGFFファイルを用意してください。
2種間のシンテニーブロック可視化手順
1.TBtools を起動する
インストール時にデスクトップにショートカットを作成していれば、そこをダブルクリックするだけでOKです。
ホーム画面には、なぜか偉人の名言がランダムで表示されます。今回はキング牧師でした。
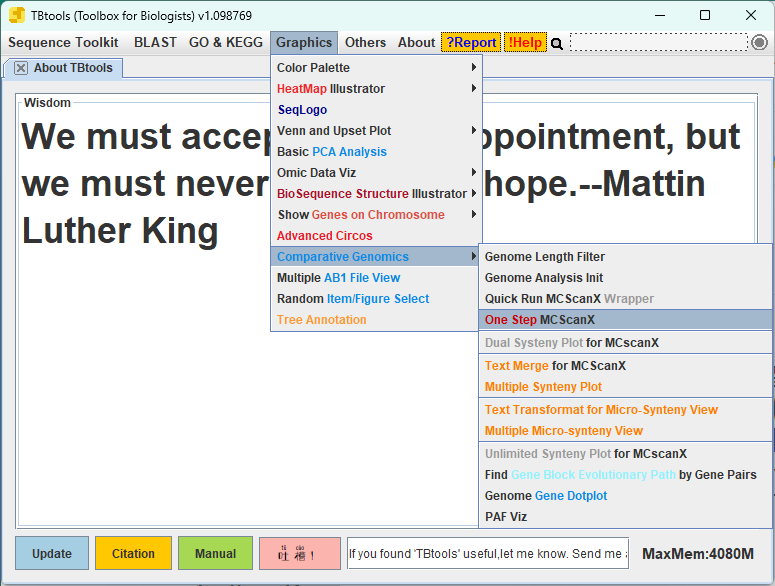
2.Graphics > Comparative Genomics > One Step MC ScanX を選択する
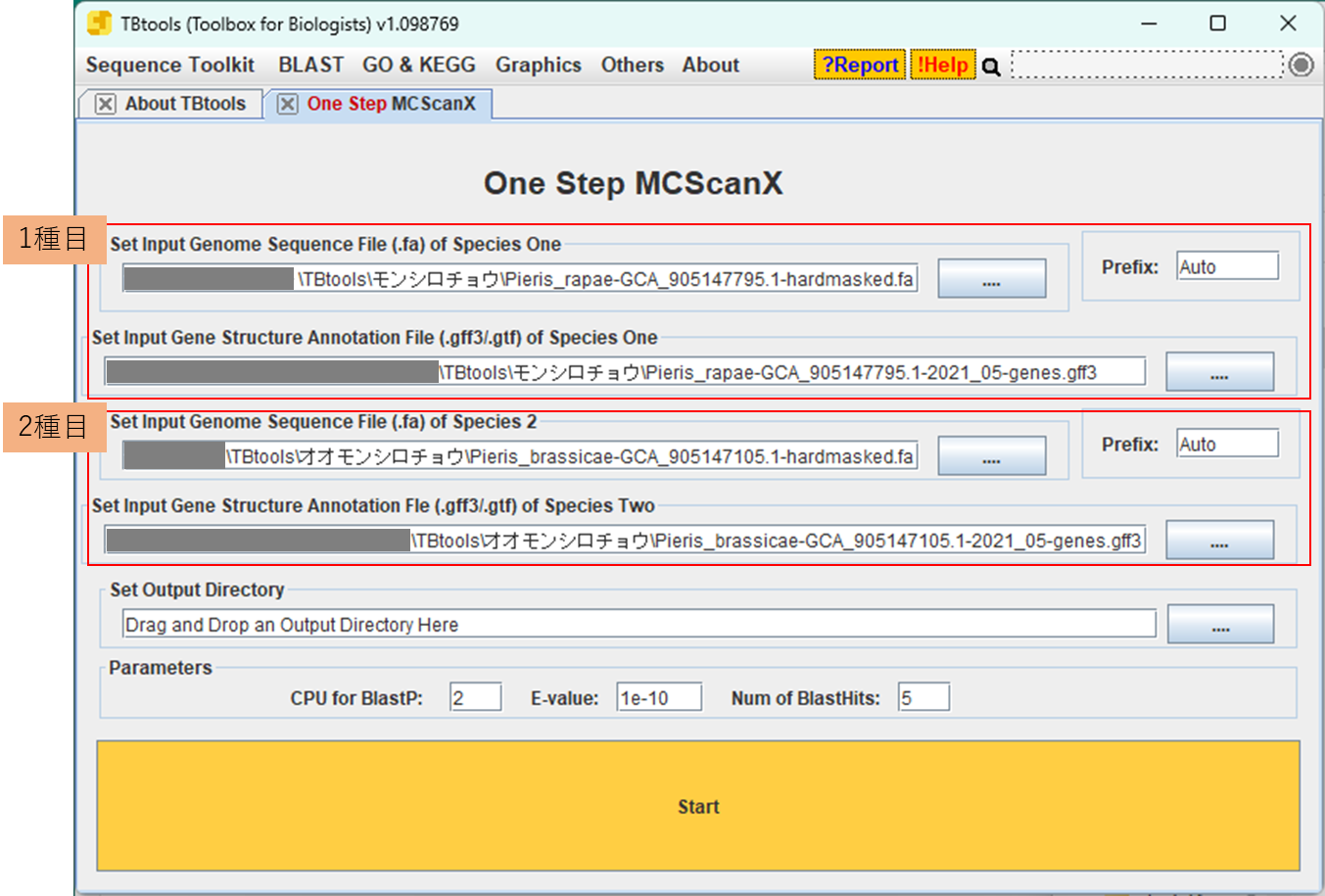
3.FASTAファイルとGFF3ファイルのパスを設定する
使いたいファイルをドラック&ドロップすればいいです。
テキストボックスの右のボタンから設定することもできます。
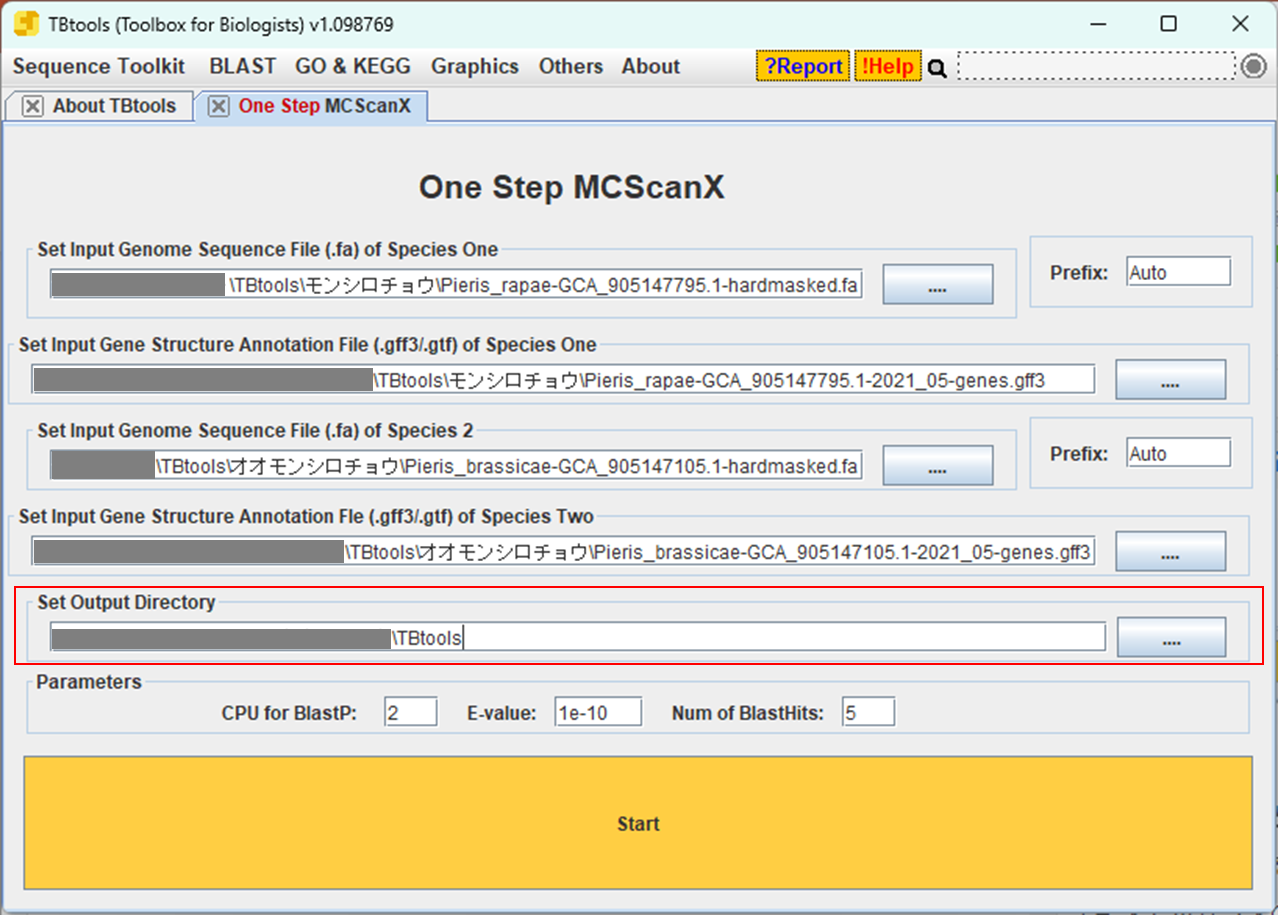
4.出力先のパスを設定する
出力先にしたいディレクトリをドラック&ドロップすればいいです。
テキストボックスの右のボタンから設定することもできます。
5.start ボタンを押す
ボタンを押下後、start の文字がグレーになってから結構長い間待ちます(シーケンスデータのサイズにもよりますが)。
フリーズしたと勘違いして終了などしないように気を付けましょう。
モンシロチョウ vs オオモンシロチョウの場合、約15分待ちました。
E-valueなど、パラメータを設定して実行することもできます。
無事実行が完了すると「MCScanX Running Finished...」というポップアップが表示されます。
出力先ディレクトリに複数のファイルが生成されているはずです。
6.Graphics > Comparative Genomics > Dual Synteny Plot for MCScanX を選択する
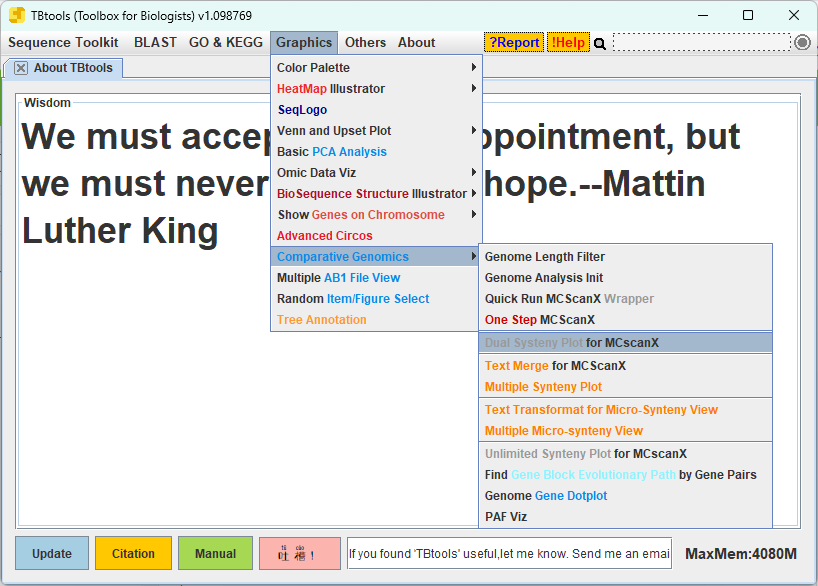
7.出力されたファイルから、拡張子が「.ctl」「.gff」「.collinearity」のファイルをそれぞれ選んで設定する
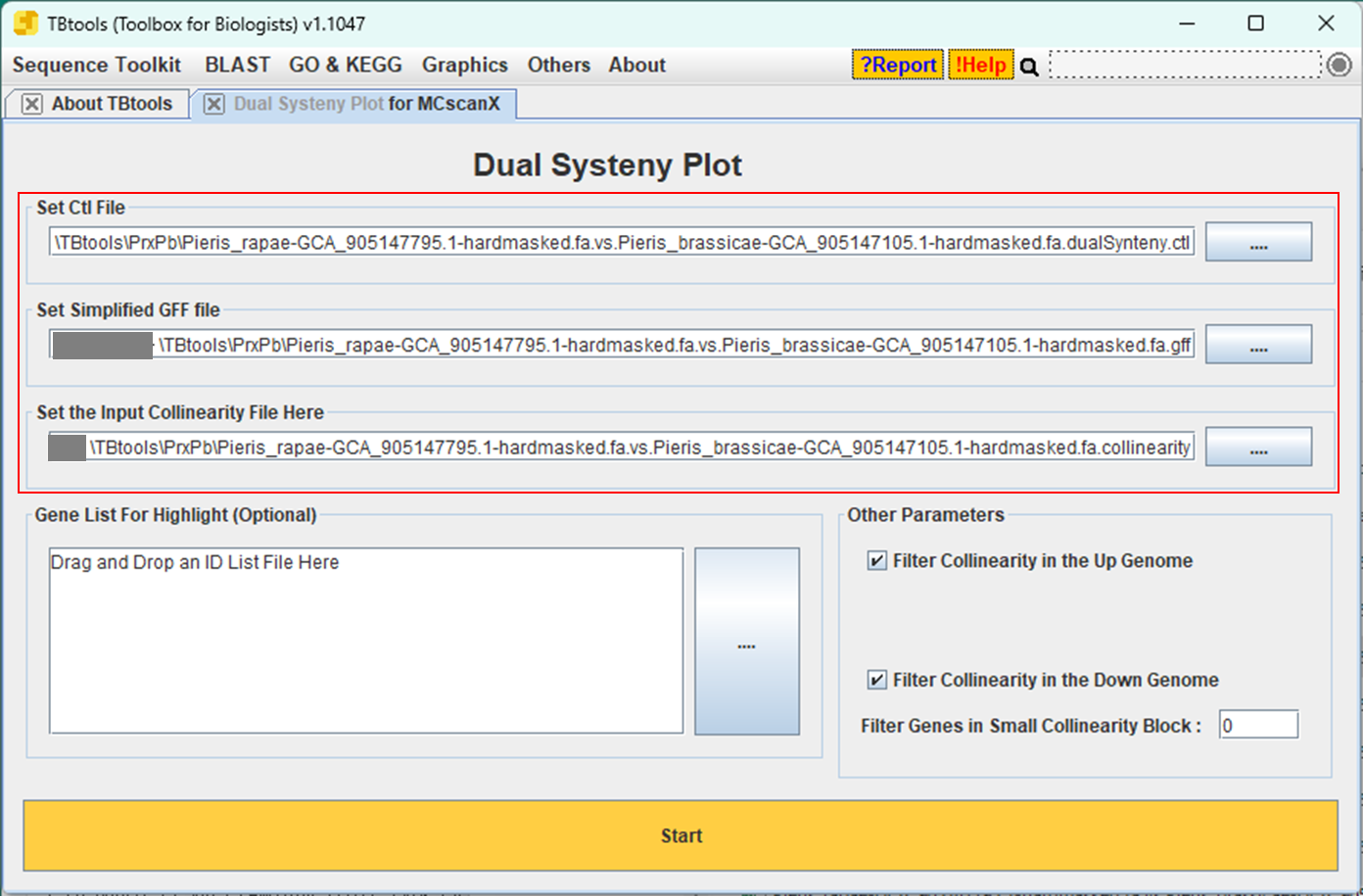
ここも同様に、出力先にしたいディレクトリをドラック&ドロップすればいいです。
テキストボックスの右のボタンから設定することもできます。
ハイライトしたい遺伝子リストがあればここで設定できます(オプション)。
8.start ボタンを押す
結果がすぐに画面に表示されます。
結果
こんな感じで画面上に結果が表示されます。
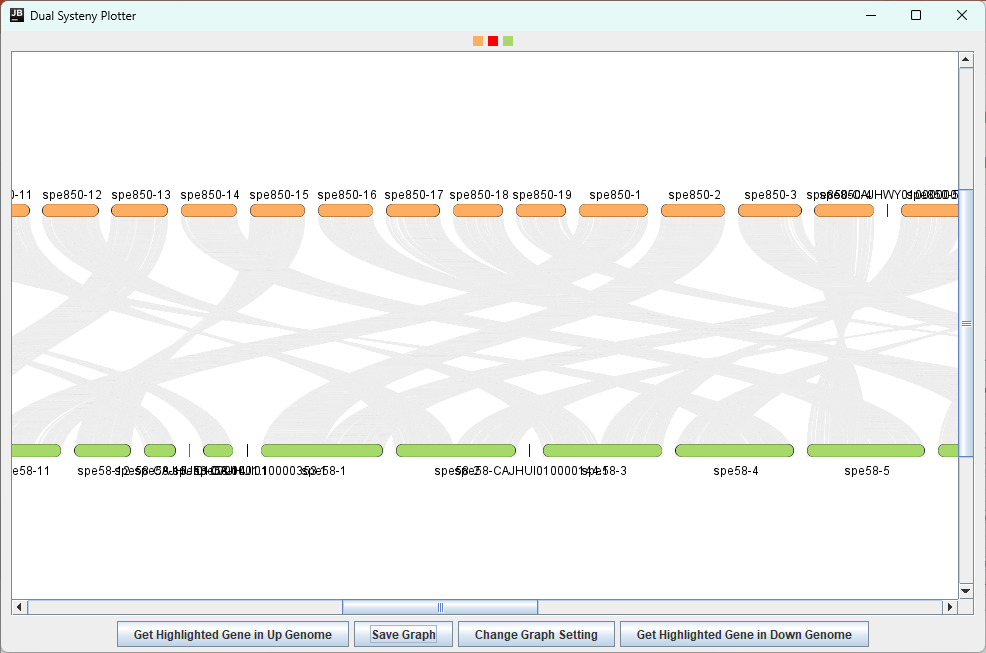
Save Graph から、.png .jpg .svg .pdf で好きなフォーマットで結果を保存できます。
拡大縮小も可能ですので、いい感じに調整して出力しましょう。
こんなときは
解析時、下記のようなエラー表示が出ることがありました。
java.io.IOException: Chr IDs is NOT consistent in Species [ファイル名]
筆者の場合、解析に使用したファイルのフォーマットが不適切な場合に本エラーが発生しました。ファイルの中身や拡張子をご確認ください。
おわりに
今回はわかりやすさのために、シンテニープロットのみにフォーカスして記事を書きました。
シーケンス技術の発展とともに分析ツールも多数出現してきておりますが、やはり非情報系生物学者には、わかりやすいことが第一ですね。
TBtoolsは他にも便利な機能が多数搭載されていますので、まずは遊んでみてください!
参考
Ensembl Rapid Release
HTSデータを扱う様々なツールをGUIインターフェースで統合した TBtools
ゲノム間のシンテニー領域を調べる MCScanX
染色体イデオグラムや染色体間のシンテニープロットを描画する RIdeogram
Genome wide study Part 14 | How to do Synteny analysis between two Species genome using TBTool
Synteny Analysis in TBTOOLS