2020年11月01日02時、それは起こった
今からちょうど一ヶ月前くらいのことです。とある奇妙なネットニュースが私の目に留まりました。
90年代のスーパー戦隊『ギンガマン』が謎のトレンド入り 複雑すぎる理由に驚愕(ニュースサイトしらべぇ)
日曜朝のネットの話題といえばスーパー戦隊やプリキュア、仮面ライダーといった「ニチアサ」があがりやすいが、「全日本大学駅伝」で潰れてしまった1日の朝。
しかし、ツイッターのトレンドには90年代のスーパー戦隊「ギンガマン」の文字が。その理由に驚きの声が相次いでいる。
■ギンガマンとは
トレンドにいたギンガマンとは、『星獣戦隊ギンガマン』のことで、1998年に放送されたスーパー戦隊シリーズの1つである。その名の通り、銀河・宇宙を意識されたキャラクターデザインとなっている。しかし、トレンド入りした理由は、ギンガマンが再放送されたり、ギンガマンの続報が出たというわけではない。
■ギンガ算のギンガ時間
トレンド入りしたのは「ギンガ時間」が訪れたからであるという。ギンガ時間は、「2020年11月01日02時30分」「2020年11月01日02時40分」「2020年11月01日02時50分」だという。
ギンガ時間はギンガ算という昔ネットで生まれた難解な覆面算から出される。日付を「ABAB年CC月BC日BA時DB分」と表記し、「A=ガ、B=ン、C=ギ、D=マ」とする。**年月日時間といった要素を抜くと「ガンガンギギンギンガマン」となる。**要するに、ネットで大昔に言われていた時間がついに現実になったことで、一部ファンが歓喜しているということだろう。
ちなみに、ギンガ算もギンガ時間も作中に登場したものでなく、ネット民が生んだものである。
ちょっとこれだけ読んでも意味を掴みかねるかと思います。記事に書いていないことを念のために補足しておくと、「ガンガンギギンギンガマン」とは、「星獣戦隊ギンガマン」のOP曲に繰り返し現れるフレーズです(実際の歌詞の表記は「ガンガンギギーン ギンガマン!」)。
だから何?
「202011010230」を「ガンガンギギンギンガマン」に読み替えられるからといって何だというのでしょう。時刻が星獣戦隊ギンガマンのOPの歌詞になるからといって何が嬉しいのでしょう。Twitterのトレンドに入るほどのことなのでしょうか?
たとえば、地上波でジブリ映画の傑作「天空の城ラピュタ」の放映があるたび、「バルス!」のシーンでTwitterのサーバーが落ちる現象は以前から知られています。「ギンガ時間トレンド入り」も同種の現象と思われるかもしれません。
しかし、ジブリ映画に匹敵するほどギンガマンのファンがインターネットに多いのかというと、そういうわけでもありません。歴代戦隊モノの人気投票ランキングを見ても、星獣戦隊ギンガマンは18位や40位に留まっており、とりたてて人気がある作品のようには見えません。実際、ギンガ時間前後のTwitterを検索してみても、ただ純粋にギンガ時間の到来を喜ぶだけで、ギンガマンの内容について語っている人はごく少数です。
おそらく、もはやギンガマンという作品そのものからかけ離れて、「ガンガンギギンギンガマン」というフレーズだけが独り立ちし、なんとなくネットミーム化しているというのが正しい現状認識ではないでしょうか。
先祖代々から村に伝わる奇妙な風習、しかしその由来を知る人間はもはや数少ない……という稗田礼二郎が遭遇するパターンによく似ています。
ギンガ算の奥深さ
さて、結局「ガンガンギギンギンガマン」というフレーズがネット民に尊ばれている理由はよくわかりませんでしたが、ギンガ算について調べるうち、だんだんその奥深さについて私は認識を改めていきました。
おさらいしておくと、ギンガ時間の算出にも用いられたギンガ算とは、**「4種類の記号(仮にA, B, C, Dと置く)を使って『ABABCCBCBADB』という12文字の配列を作り、ひと目見ただけでは『ガンガンギギンギンガマン』と言っているようには見えないようにする遊び」**のことです。
上記のニュースやニコニコ大百科の記事ではこれを覆面算と呼んでいますが、そもそも等式を作ることを目的にはしていないため、正確には覆面算とは呼べません。むしろ、古代ローマの執政官ユリウス・カエサルが使ったとされる単一換字式暗号の一種と言えるでしょう。
A, B, C, Dというアルファベットでギンガ算の例を出しましたが、暗黙のルールとして、ギンガ算に使われる記号は文字ではなく図形でもいいようです。実際にギンガ算を収集している謎のサイト「KOKO.TTO.TOKYO」から、個人的に美しいと思ったギンガ算の図形の例を5つ引用して、以下にお示しします。

いかがでしょうか? これらの幾何学図形を何の文脈もなくひと目見て、「1998年に放送されたスーパー戦隊の主題歌を歌っている!」と気付ける人類は果たしてどれくらいいるのでしょうか?
念のため、上の図形でそれぞれどの部分が「ギ」「ン」「ガ」「マ」に対応しているのかのヒントを添えた画像を作成したので、下に貼っておきます。
しかしできることならば、これらのヒントを見ることなく、ご自分で上の図形をじっと眺めて、その背後に隠されたルールに気付き、私と同じ感動を味わって欲しいです。
**ヒントを見るにはここをクリック**
もはや狂気の沙汰ですが、やはり一種の美しさを感じることは否めません。
ギンガ時間の謎を追求していくうちにギンガ算そのものに魅了された私は、やがて自分でもギンガ算を作りたいという欲求が芽生え始めました。既にギンガ算を発表するのに絶好のタイミング(ギンガ時間、次は3030年)は過ぎてしまいましたが、ワケあって今日こうしてQiitaの**「バイオインフォマティクス」**アドベントカレンダーの記事として投稿することにしました。
そのワケとは……この記事のタイトル、そしてギンガ算を構成する記号が**「4種類」**であることから察して頂けるかと思います。
ギンガ遺伝子、あるいはギンガモチーフ

そう、DNA(デオキシリボ核酸)です。
あらゆる生物はDNA配列に遺伝情報を格納しています。DNAはデオキシリボース・リン酸・塩基の三つの部位から構成される化学物質です。このうち塩基は、アデニン(A)・チミン(T)・グアニン(G)・シトシン(C)の4種類が存在します。よって、あらゆる生物の遺伝情報はA, T, C, Gの4種類の文字のみで表現することができます。
そして4種類の文字があれば、ギンガ算も表現することができるというワケです。
もっとも、インターネットの集合知を見くびってはいけません。この程度のことはギンガ算に対する情熱と高校生物程度の知識があれば誰でも思いつくはずです。
考古学的調査の結果、ギンガ遺伝子という概念は、遅くとも2017年には既に提出されていたことが確認できました。
したがって、DNAでギンガ算を表現するというアイデア自体にはオリジナリティはありません。
なお、生物学的に厳密に言うと、すべてのDNA配列が遺伝子と呼ばれるわけではなく、タンパク質をコードしている塩基配列のことのみを指して**「遺伝子」**と呼ばれます。
**"ATATGGTCTACT"**のように短い配列は、遺伝子というよりは、モチーフと呼ばれることが多いように思います(※)。
以下では厳密化のために、ギンガ算となる塩基配列を「ギンガ配列」あるいは「ギンガモチーフ」、**ギンガ配列を含む遺伝子を「ギンガ遺伝子」**と呼んで区別することにしましょう。
(※) 実際にモチーフと呼ばれるには、頻繁に現れる配列であったり、生物学的に意味のある配列である必要があります。
明らかに、ギンガ配列は"ATATGGTCTACT"だけではありません。「ギ」「ン」「ガ」「マ」のどれに「A」「T」「G」「C」を割り当てるかによって、 $ 4!=24 $ 種類のギンガ配列が存在します。
Mathematicaでは、次の1行のコマンドですべてのギンガ配列を記述することができます:
# 1 <> #2 <> #1 <> #2 <> #3 <> #3 <> #2 <> #3 <> #2 <> #1 <> #4 <> #2 & @@@ Permutations[{"A", "T", "G", "C"}]
結果は以下のようになります:
{ATATGGTGTACT,ATATCCTCTAGT,AGAGTTGTGACG,AGAGCCGCGATG,ACACTTCTCAGC,ACACGGCGCATC,TATAGGAGATCA,TATACCACATGA,TGTGAAGAGTCG,TGTGCCGCGTAG,TCTCAACACTGC,TCTCGGCGCTAC,GAGATTATAGCA,GAGACCACAGTA,GTGTAATATGCT,GTGTCCTCTGAT,GCGCAACACGTC,GCGCTTCTCGAC,CACATTATACGA,CACAGGAGACTA,CTCTAATATCGT,CTCTGGTGTCAT,CGCGAAGAGCTG,CGCGTTGTGCAG}
さて、中立進化と言って、ゲノム(ひとつの生物の全DNA配列)のかなりの部分が、偶然の突然変異から生み出されたランダムな配列であると言われています。
4種類の文字から成る長さ12文字の配列は、全部で $4^{12}$ 通りあります。よって、12塩基長のランダムなDNA配列があれば、それは$4!/4^{12} = 1.43 * 10^{-6}$の確率で**「ギンガ配列」**となります。
日常的な感覚からすると、1%よりもずっと小さいこの数字はとても低い確率に見えます。しかしヒトゲノムのサイズは3億 $(3*10^8)$ 塩基対もあります。ヒトはゲノムの5%しか遺伝子ではないと言われているので、いっそすべてがランダムな配列だと仮定してざっくり試算すると、
\frac{3*10^{8}}{12} * \frac{4!}{4^{12}} \approx 358
358個ものギンガ配列がヒトゲノムの中に存在することが予想されます。
つまり、単にヒトゲノムの中にギンガ配列を求めても、それは確率的に「あって当然」なので、有り難み/面白み/美しさに欠けてしまいます。美しさはギンガ算の必要条件です。これではまだ「ギンガ算を作った」とネット民たちは認めてくれないでしょう。
ヒトゲノムにおけるギンガ遺伝子の探索
そこで、単なるギンガ配列ではなく、ギンガ遺伝子を求めることにしましょう。ギンガ遺伝子のナイーブな定義は上述したように「ギンガ配列を持った遺伝子」です。ここではもうひとつ条件を加えて、
・5’→3'方向でギンガ配列を含んでいる
ということを要求しましょう。
DNAの相補的な二本鎖は「ワトソン鎖」「クリック鎖」と呼ばれて区別され、それぞれ読み取られる方向は逆向きになっていますが、遺伝子はどちらの鎖にもコードされ得ます。つまり、「ワトソン鎖の5’→3'方向ではギンガ配列になっているが遺伝子にはなっておらず、クリック鎖の5’→3'方向ではギンガ配列になっていないが遺伝子にはなっている」というケースも存在します。今回の解析ではそのような美しくないケースを排除しましょう。
果たしてこのような遺伝子は私たち人間の中にどれくらい存在しているのでしょうか?
これを調べるため、まずは**NCBI(アメリカ国立生物工学情報センター)**の提供するホモ・サピエンスの全遺伝子の配列情報(transcript)をダウンロードします。
この配列情報はA, T, G, CとDNAの記号で書かれていますが、実際はスプライシング後のRNAの配列になっています。スプライシングとは、DNA→(転写)→RNA→(翻訳)→タンパク質というセントラルドグマの中で、真核生物だけで行われる転写後の現象で、イントロンと呼ばれる領域を切り捨て、エクソンと呼ばれる領域だけを残して繋ぎ直す、「継ぎ接ぎ作業」のことです。
配列解析を行うソフトは色々ありますが、今回はBiopythonを使っていきます。使ったモジュールは以下の通りです。
import pandas as pd
import re
from Bio import Entrez, SeqIO
from Bio.Seq import Seq
Entrez.email = "xxx@xxxx.com" # NCBIに伝える自分のメールアドレスをここに記入しておく。
ダウンロードしたFASTAファイル(配列情報でよく使われるファイル形式)を読み込んで、まずは全遺伝子の配列情報を格納したデータフレームを作成します。
records = SeqIO.parse ('GCF_000001405.39_GRCh38.p13_rna.fna', 'fasta')
ids = []
descs = []
seqs = []
for record in records:
ids.append(record.id)
descs.append(record.description)
seqs.append(record.seq)
RNAs = pd.DataFrame()
RNAs["id"]=ids
RNAs["desc"]=descs
RNAs["seq"]=seqs
RNAs = RNAs.set_index("id")
# reを使うために、配列情報のコラムをstr化しておく。
RNAs["strseq"] = [str(seq) for seq in RNAs["seq"]]
次に、Mathematicaで作成した24種類のギンガ配列を格納したファイル(permutation.txt)を読み込み、これを各遺伝子の配列情報と比較し、それぞれの遺伝子がギンガ配列を含んでいるか否かを判定します。
# ギンガ配列の読み込み
gingaseqs = pd.read_table("permutation.txt")
# 各ギンガ配列に対して、各遺伝子がその配列を含んでいるかをTrueFalseで返す
gingaTFs = [
[re.search(gingaseq, strseq) is not None for strseq in RNAs["strseq"]]
for gingaseq in gingaseqs["strseq"]
]
# いずれかのギンガ配列を含んでいる遺伝子のみのデータフレームを作成する
gingadfs = [RNAs[gingaTF] for gingaTF in gingaTFs]
for i in range(24):
#その遺伝子がどのギンガ配列を含んでいるかを"gingaseq"列に記しておく
gingadfs[i]["gingaseq"] = gingaseqs["strseq"][i]
gingadfs = pd.concat(gingadfs)
# ギンガ配列を含む遺伝子のindexの集合を作る
gingaindexs = set()
for gingaTF in gingaTFs:
gingaindexs = gingaindexs|set(RNAs[gingaTF].index)
# ギンガ配列を含む遺伝子の数
print(len(gingadfs))
こちらの実行結果は意外にも**「645」**となり、先程の見積もりよりもむしろ数が多いです。これは元のファイルにスプライシングバリアント(スプライシングによって同じゲノム領域から生まれる遺伝子の亜種)が含まれているため、ゲノム上の同じギンガ配列を何度もカウントしてしまっていることが原因のひとつでしょう。
これだけギンガ配列を含む遺伝子の数があるということは、ギンガ遺伝子の定義にもう少し厳し目の条件を要求してもいいということです。美的感覚を満たすべく、どんどん条件を課していきます。次に加える条件はこちらです。
・コーディング領域にギンガ配列がある
スプライシング後の成熟mRNAでも、すべての配列がタンパク質として翻訳されるわけではありません。タンパク質のコーディング領域の5'側と3'側のそれぞれに非翻訳領域(UTR)が存在します。ここにある配列はイントロンと同じように、「最終的に捨てられる」情報と言っていいでしょう(※)。そんなところにギンガ配列を持っている遺伝子をギンガ遺伝子と呼ぶのは躊躇われます。
(※)実際には何らかの機能を持っている場合もあります。
RNAからタンパク質のコーディング領域(通称CDS)のみを抽出するために、efetch関数を使ったマイ関数を作っておきます。efetchがどのような引数を使えるかは、NCBIの次のページが参考になりました:
https://www.ncbi.nlm.nih.gov/books/NBK25499/table/chapter4.T._valid_values_of__retmode_and/
# mRNAのコーディング領域のみを抽出する関数
def CDSextract(mRNAid):
handle = Entrez.efetch(db="nuccore", id=mRNAid, rettype='fasta_cds_na', retmode='text')# , retmode='text'
record = handle.read()
handle.close()
searchresult = re.search("CDS](.*)", record.replace("\n",""))
if searchresult is not None:
returnvalue = searchresult[1]
else:
returnvalue = "None"
return returnvalue
この関数を使って、先程作った「ギンガ遺伝子候補」のデータフレームに、CDSの情報を付加していきます。
# すべてのギンガ遺伝子候補のCDSを取得する ※時間かかる
CDSs = [CDSextract(mRNAid) for mRNAid in gingadfs.index]
gingadfs["CDS"] = CDSs
# CDSがデータベースにあったものだけをデータフレームに残す
CDSgingadf = gingadfs[gingadfs["CDS"]!="None"].copy()
# CDSにギンガ配列が含まれているかどうかを判定する
CDSginga = []
for RNAid in CDSgingadf.index:
result = re.search(CDSgingadf.loc[RNAid]["gingaseq"], CDSgingadf.loc[RNAid]["CDS"])
if result is not None:
CDSginga.append(result.span() )
else:
CDSginga.append("None")
# ギンガ配列が含まれているか否かの列名は"CDSginga"
CDSgingadf["CDSginga"] = CDSginga
# CDSにギンガ配列が含まれているものだけを残す
TrueCDSgingadf = CDSgingadf[CDSgingadf["CDSginga"]!="None"].copy()
# CDSにギンガ配列が含まれている遺伝子の数
print(len(TrueCDSgingadf))
こちらの実行結果は287個と、まだまだ多いです。もっと条件を要求していきましょう。次なる条件として考えるのはこちらです。
・コドンの読み取り枠がギンガ配列と一致している
タンパク質はアミノ酸の配列から構成されていますが、1つのアミノ酸は連続する3つの塩基から決定されています。この3個1組の塩基のことをトリプレット・コード、コドンと呼びます。12文字からなるギンガ配列はちょうど3の倍数なので、コドンの読み取り枠にぴったり収めることができます(ここ美しポイント)。つまり、「ガンガ(ATA)」「ンギギ(TGG)」「ンギン(TGT)」「ガマン(ACT)」というような、4つのコドンから4つのアミノ酸が作られるようなギンガ配列だと美しいと言えるでしょう。逆に、読み取り枠がずれて、「XAT」「ATG」「GTG」「TAC」「TXX」と、ギンガ算以外の任意の文字Xを含んで翻訳されるような遺伝子は、美しいとは言えません。
この条件は、CDS内におけるギンガ配列の開始位置が、ちょうど3で割り切れるかどうかによって判定できます。
# ギンガ配列がリーディングフレームに一致しているかどうかを判定する
gingacodon = [gingaspan[0]%3 == 0 for gingaspan in TrueCDSgingadf["CDSginga"]]
# ギンガ配列がリーディングフレームに一致しているか否かの列名は"gingacodon"
TrueCDSgingadf["gingacodon"] = gingacodon
# ギンガ配列がリーディングフレームに一致しているかものだけを残す
Codonmatchdf = TrueCDSgingadf[TrueCDSgingadf["gingacodon"]==True].copy()
# 遺伝子名の列を作っておく
genename = [re.search("\((.*)\)", desc)[1] for desc in Codonmatchdf["desc"]]
Codonmatchdf["genename"] = genename
# ギンガ配列がリーディングフレームに一致している遺伝子の数
print(len(Codonmatchdf))
こちらの実行結果は79個と、だいぶギンガ遺伝子の候補を絞り込むことができました。しかしまだまださらなる条件を要求することができます。次に要求したい条件はこちらです
・実験的に存在が確認されている
NCBIからダウンロードしたファイルには、ヒトの細胞を使った実験によって存在が確認された遺伝子だけでなく、他の生物種の配列との相同性などから理論的に存在を推定された遺伝子も含まれています。そのような遺伝子はIDのプレフィックスがXMとなっています。存在があやふやなものには今は興味がありません。確かに存在するギンガ遺伝子だけを見ていきましょう。
# XMかどうかを判定する
predicted = []
for RNAid in Codonmatchdf.index:
result = re.search("XM", RNAid)
if result is not None:
predicted.append("Predicted")
else:
predicted.append("Exist")
Codonmatchdf["predicted"]=predicted
# 確実に存在するものだけを残す
Existdf = Codonmatchdf[Codonmatchdf["predicted"]=="Exist"].copy()
# ファイル出力
Existdf.to_csv("exist.csv")
# 実験的に存在が確認されたギンガ遺伝子の数
print(len(Existdf))
こちらの実行結果は19個となりました。そろそろデータフレームの中身を見てみましょう。
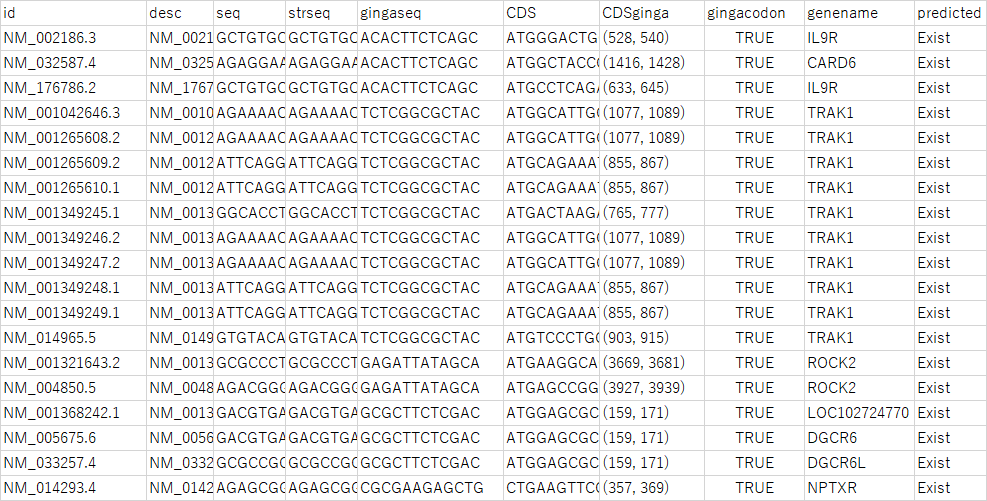
おや? 同じgenenameがいっぱい並んでいます。descriptionを見てみると、やはりスプライシングバリアントが多く含まれていることがわかります。LOC102724770とDGCR6Lもdescriptionを見ると、DGCR6のバリアントらしいことがわかりました。結局のところ、
- ギンガ配列を含む遺伝子である
- 5’→3'方向でギンガ配列を含んでいる
- コーディング領域にギンガ配列がある
- コドンの読み取り枠がギンガ配列と一致している
- 実験的に存在が確認されている
という5つの条件を満たす真のギンガ遺伝子は、実質的に以下の6つのみ、ということになります。
| ギンガ遺伝子 | 含まれるギンガ配列 | 関連する機能 |
|---|---|---|
| IL9R | ACACTTCTCAGC | 免疫 |
| CARD6 | ACACTTCTCAGC | アポトーシス |
| TRAK1 | TCTCGGCGCTAC | 細胞内輸送 |
| ROCK2 | GAGATTATAGCA | 細胞骨格の制御 |
| DGCR6 | GCGCTTCTCGAC | 心血管形成 |
| NPTXR | CGCGAAGAGCTG | シナプス再編成 |
遺伝子名をクリックすると**KEGG(京都遺伝子ゲノム百科事典)**の各遺伝子の全塩基配列を載せているページへのリンクへ飛びます。是非リンク先で、右の配列をページ内検索して、確かに引っかかることを確認してみてください(TRAK1とNPTXRは改行があるせいで検索に引っかかりにくいことに注意です)。
ROCK2はギンガ遺伝子である
上記の6つの遺伝子のリストを見たとき、おそらく生物学者が最も関心を引かれるのは、ROCK2でしょう。
こちらのROCK2はROCK (Rho-associated coiled-coil-containing protein kinase)ファミリーの2番目の遺伝子で、細胞骨格の制御という細胞の根本的な機能に関わっているため、そこから派生する数多くの生命現象──細胞運動・細胞極性・細胞接着・細胞分裂・アポトーシス・転写制御・神経管形成・神経突起伸展制御・シナプス形成・シナプス可塑性──に関与しており、非常に数多くの研究がなされています。
その重要さを端的に示す証拠として、ROCKの機能を解説する日本語のページまで存在しています。
ヒトの遺伝子は約2万個ありますが、その中でも日本語で解説記事が書かれる遺伝子はごく少数です! ROCK2が具体的にどのような役割を細胞内で果たしているかに興味がある方は、是非この脳科学辞典の記事を参照してみてください。
さて、ギンガ配列がROCK2のコーディング領域に含まれているということは、ギンガ配列から翻訳された4つのアミノ酸がROCK2タンパク質のどこかに含まれています。具体的に言うと、タンパク質のN末端から数えて1310-1313番目がそうです。果たして、このギンガ配列から翻訳された領域はROCK2においてどのような役割を果たしているのでしょうか?
幸運なことにROCK2はよく研究されているので、細胞内でどのような立体構造を取り、どのような細胞内局在をするかまで大体わかっています。**Linda Truebestein, Daniel J. Elsner & Thomas A. Leonard (2016) Made to measure – keeping Rho kinase at a distance**によくまとまった図があったので、こちらを引用させていただきます。
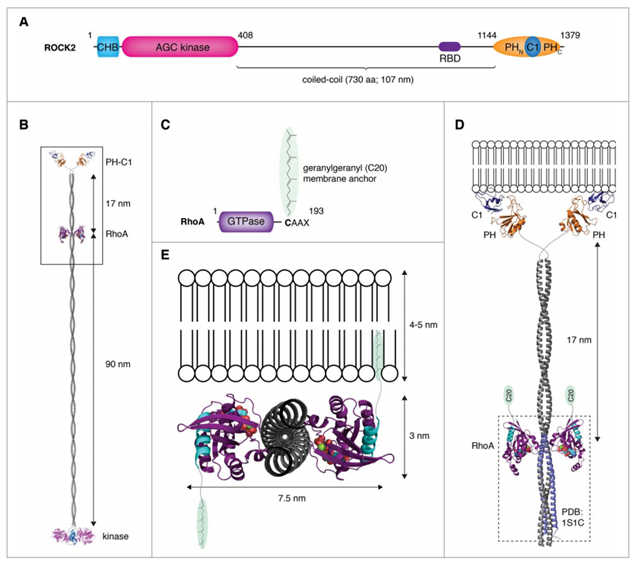
1310-1313のアミノ酸は、上図を見るとわかるように、ROCK2の中でもPHドメインのC末端側に位置することがわかります。PHドメインは非常に有名な機能性モチーフで、細胞膜に結合する機能を持ち、タンパク質を細胞膜近辺に局在させる役割を持っています。ということは、PHドメインに含まれるギンガ配列もまた、ROCK2を細胞膜近辺に留めておく役割に貢献している、ということができるでしょう。
おわりに
ROCK2という超有名な遺伝子がギンガ遺伝子であることが判明したとき、私は得も知れぬ快感に襲われました……。「そうか、本当に私を構成する細胞のひとつひとつに、ギンガ配列を含むギンガ遺伝子が存在し、そしてギンガ遺伝子から翻訳されたギンガタンパク質が存在するのだな……私が自分の声帯で歌うことを覚えるよりも早く、既に私を構成する細胞たちは星獣戦隊ギンガマンのオープニングテーマを歌っていたのだな……」と実感できた瞬間です。
だから何?