はじめに
この前、ひさしぶりにPythonでデータ解析したんです。
pandasの使い方とかめちゃくちゃ忘れてたんですが。
そのとき、ふと思った事がありまして、
「そういえばうちの学科の同期でPython使ったことある人、あんまり聞いたことないな」と。
Rの使い手は何人かちらほら聞くんですけどね...
(簡単やし、パッケージも多いし当たり前か、という感じです。)
Pythonは化学も出来るんやぞということを紹介します。
※packageのインストールとかは省いてます。
RDkit
Cheminformatics(化学情報学)分野で大活躍するpackageです。
化合物を描画したり、構造データを読み込んでグラフ構造を見たりします。
RDkitでできること→https://www.rdkit.org
描画
まずは適当に構造を入れてみましょう。
from rdkit import Chem
from rdkit import IPythonConsole #構造描画の設定
from rdkit.Chem import Draw
ここで構造の入力にはSMILESという記法を使います。
SMILESはSimplified Molecular Input Line Entry Systemの略で、構造情報を文字列に変換して書く記法です。
詳細はこちらを見てください(http://opensmiles.org)
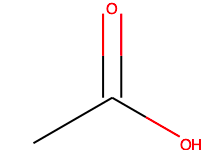
例えば酢酸とか描画してみますか。
m1=Chem.MolFromSmiles("CC(=O)O") #smileで書いた文字列を読み込むメソッド
m1
すると酢酸の構造式が描画できちゃう、なんて簡単。
<出力結果>
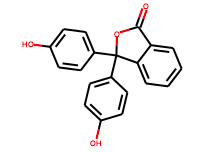
同様にフェノールフタレインみたいな芳香族化合物も描画出来る。
m2=Chem.MolFromSmiles("O=C1OC(c2ccccc12)(c3ccc(O)cc3)c4ccc(O)cc4")
m2
<出力結果>
データからとってくる
例えばデータセットを作って解析したいな、とか思ったりしたとき。
データベースとして紹介するとすれば、
●ChEMBL(https://www.ebi.ac.uk/chembl/)
●PubChem(https://pubchem.ncbi.nlm.nih.gov)
このあたりが面白いと思います。
NIHのサイトが好きなので今回はPubChemを使います。
解熱作用で有名な「アスピリン」を検索すると下の検索画面が出てきます。
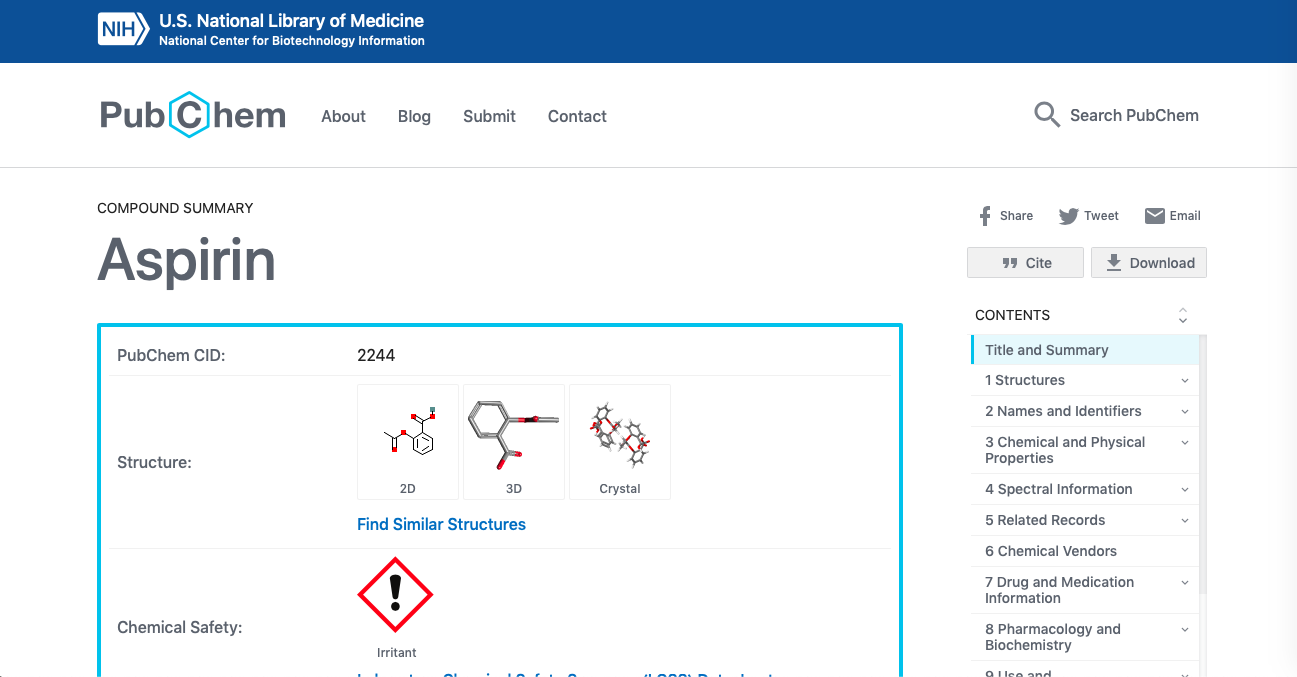
Downloadボタンから2D sructure sdfファイルをsaveしましょう。
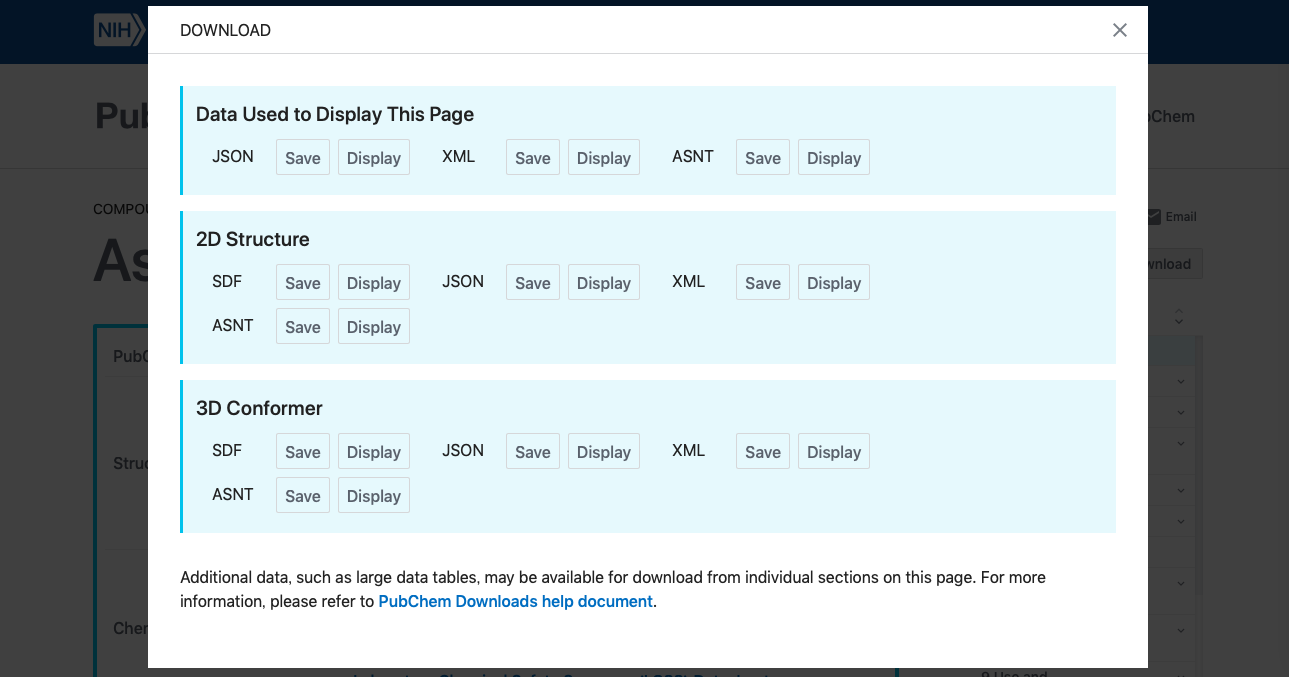
d1=Chem.SDMolSupplier("Structure2D_CID_2244.sdf")#sdfファイルを読み込む
Draw.MolsToGridImage(d1)
<出力結果>
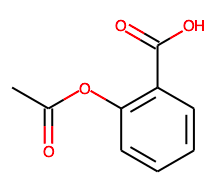
ちゃんとアスピリンが召喚されました。
構造探索
単に出力しただけで満足せず、出力した構造をいろいろいじっていきます。
探索
2種類の化学物質の部分的な構造の一致場所を見ます。
# 酢酸とギ酸の構造一致性
c1 = Chem.MolFromSmiles('CC(=O)O') #酢酸
c2 = Chem.MolFromSmarts('O=CO') #ギ酸
c1.GetSubstructMatches(c2) #酢酸とギ酸で構造が一致する部分のチェック
<出力結果>
((2,1,3))
この数字はギ酸と対応する酢酸のatom indexです。
削除
from rdkit.Chem import AllChem #分子の修正を加えるためのモジュール
delete = AllChem.DeleteSubstructs(c1,c2)# 酢酸からギ酸との共通部分を削除
Chem.MolToSmiles(delete)
<出力結果>
'C'だけ残りましたね
置換
add = Chem.MolFromSmiles('CC(O)C(=O)O') #置換する部分構造(乳酸)
exc = AllChem.ReplaceSubstructs(c1,c2,add) #酢酸とギ酸の共通部分を乳酸に置換
Chem.MolToSmiles(exc[0])
<出力結果>
存在するのかわかりませんが、'C.CC(O)C(=O)O'みたいな未知の化合物も合成できます。
類似性指標
Fingerprintという分子の類似性を示すのに必要な、化学的特徴を記述する手法があります。
# 酢酸、ギ酸、乳酸のフィンガープリントを用いた類似度計算
from rdkit import DataStructs
from rdkit.Chem.Fingerprints import FingerprintMols
data=[Chem.MolFromSmiles('CC(=O)O'),Chem.MolFromSmiles('O=CO'),Chem.MolFromSmiles('CC(O)C(=O)O')]#類似度を調べたい3分子をつっこむ
judge = [FingerprintMols.FingerprintMol(x) for x in data]
DataStructs.FingerprintSimilarity(judge[1],judge[2])#ギ酸と乳酸の類似度
<出力結果>
0.19くらいでした。見た目からしてあんまり似てないですね。
類似性を測る指標としてはTanimoto、Diceあたりが有名ですかね。
DataStructs.FingerprintSimilarityではデフォルトがTanimoto係数なので別の指標にしたければ
DataStructs.FingerprintSimilarity(fps[1],fps[2], metric=DataStructs.DiceSimilarity)#類似性指標の変更
<出力結果>
0.3125
これで、Diceによる指標となります。
類似度マップ
この類似度をマップに描画します。
例えば、安息香酸ベースにしたラクトースの類似度マップを描画します。
from rdkit.Chem.Draw import SimilarityMaps #類似度マップのモジュール
mol = Chem.MolFromSmiles('O=C(O)c1ccccc1')#安息香酸
refmol=Chem.MolFromSmiles('O[C@H]1[C@H](O)[C@H](OC(O)[C@@H]1O)CO[C@@H]2O[C@H](CO)[C@H](O)[C@H](O)[C@H]2O')#ラクトース
fp = SimilarityMaps.GetTTFingerprint(refmol, fpType='normal')#topological torsions finger print
fig, maxweight = SimilarityMaps.GetSimilarityMapForFingerprint(mol, refmol, SimilarityMaps.GetMorganFingerprint, metric=DataStructs.TanimotoSimilarity)#Tanimoto係数によるマッピング
<出力結果>
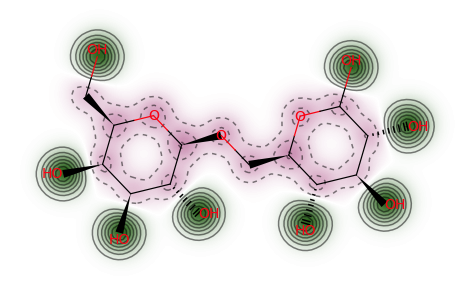
電荷による可視化
今度は安息香酸の電荷による可視化をやります。
AllChem.ComputeGasteigerCharges(mol)
contribs = [float(mol.GetAtomWithIdx(i).GetProp('_GasteigerCharge')) for i in range(mol.GetNumAtoms())] #Gasteiger法を用いた部分電荷による可視化
fig = SimilarityMaps.GetSimilarityMapFromWeights(mol, contribs, colorMap='jet', contourLines=10)
<出力結果>
電荷作用をcolor mapで可視化できました。
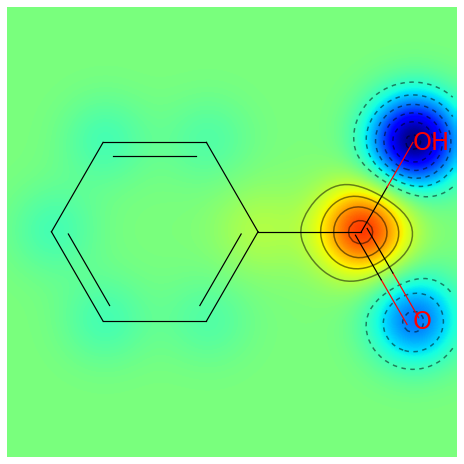
おわりに
描画から構造解析、編集までできるRDkitは便利です。
Pythonでおもしろいpackageとかあればまた試したいと思います。