NGSデータ解析プラットフォーム 「Galaxy」 のチュートリアル 「From peaks to genes」 の日本語版を作成したときに、
「ここ分からないよ!」「チュートリアル通りにならない!」
となる部分がある気がしたので、追加で説明します。
(上記の日本語版記事とともにご覧ください)
Galaxyの登録について
まずは Galaxy の登録について記事ではほとんど触れていないので、ここから説明します。
- Galaxy のトップにある ユーザー(または User )(下図の赤枠部分)をクリックし、続いて登録を選択する。
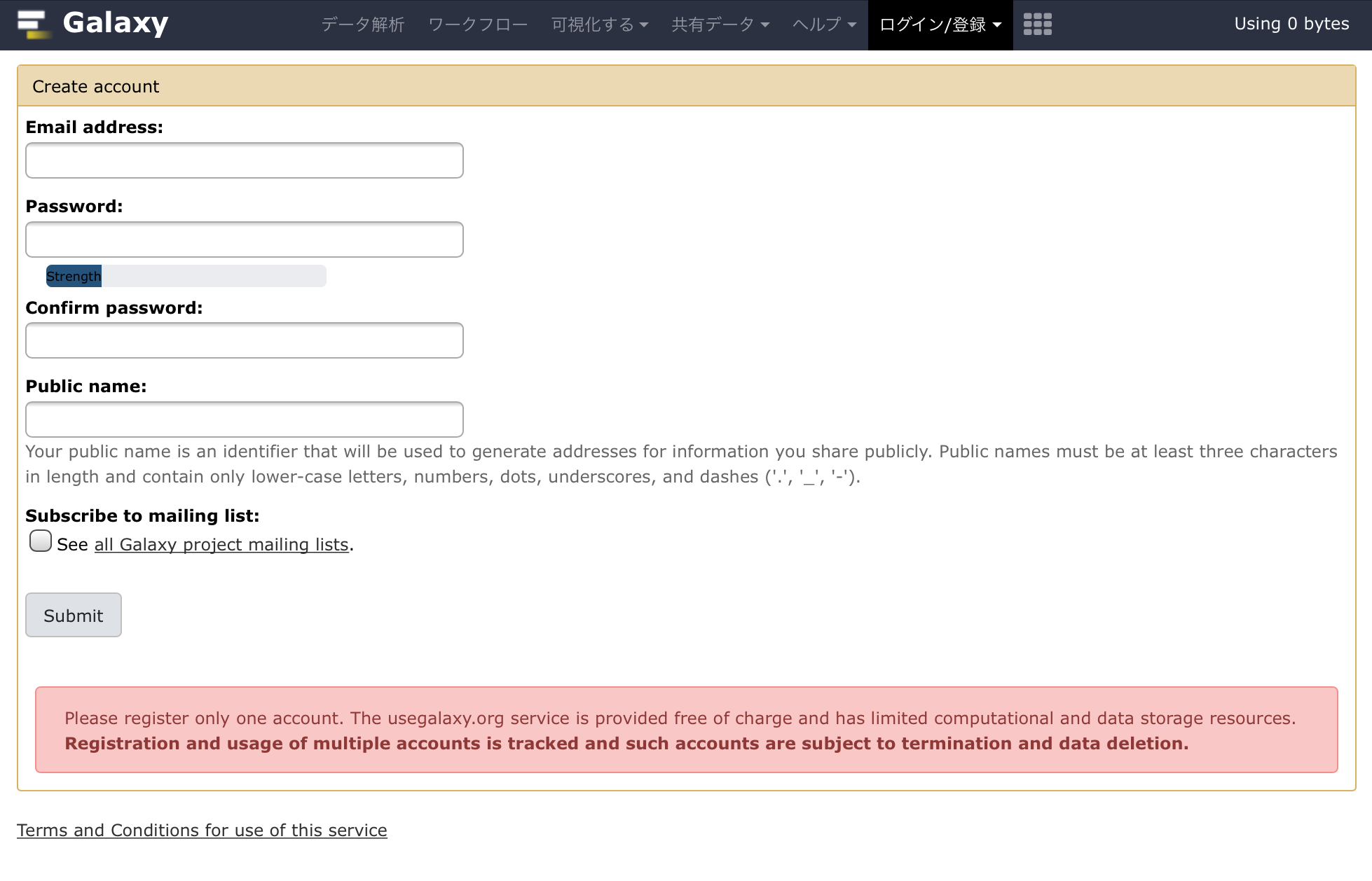
- 次の画面が出てくるので必要事項を記入して Submit をクリック。
- 入力したメールアドレスに登録確認のメールが来るので、そのメール内にあるURLをクリックする。
- 登録完了!!
ツールの再実行について
ツール Replace Text on data ... を再実行する際に「もう一度このジョブを実行する」ボタンがどこにあるかわからない人向けです。
- History パネル(右側のパネル)内にある
Replace Text on data ...をクリックして、対象のヒストリーを展開する。
- フォーマットやデータベースについて書かれている欄の下に5つのアイコンが表示されているはずです。そこにある ループ矢印 のアイコン(真ん中にあるアイコン)が再実行のボタンです。
- クリックすると中央のパネルにツールの設定画面が現れるので、チュートリアル従って各種設定を行い、再実行してください。
可視化について
(2018.06.14 時点での内容です)
Galaxy のアップデートによって、チュートリアルの記事のようにハンズオンできない状態になっています(可視化自体はできます)。
可視化に関しては、
可視化する(chart) のアイコンをクリック → 中央のパネルにて良さそうなグラフを選択 → 設定を色々いじってみる
といった手順で操作して、染色体ごとの遺伝子数が分かるような棒グラフを作成してみてください。
ワークフローの抽出について
いくつか分かりにくい点があると思うので下に箇条書きしておきます(ほかにも分かりにくい点がありましたら随時コメントください)。
- インプットしたデータセットを改名する部分。
GEO からアップロードしたピークファイルの名前をPeak regionsにして、UCSC からアップロードしたマウスの遺伝子リストをReference regionsにする。
- Auto Re-layout 後のワークフローのレイアウト
下の図のようになっていれば(恐らく)正しく作成されています。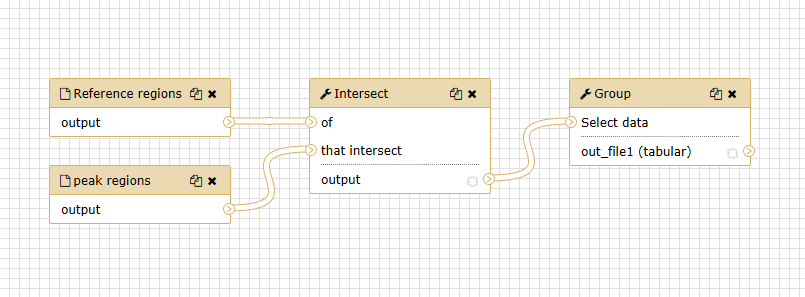
チュートリアルでは(インプットしたデータセットの名前を変えさせているくせに)、2つとも「Input dataset」のまま接続している図を載せているため、非常に分かりにくくなっています。
そのくせインプットデータの接続先を逆にすると、この後のハンズオン(ワークフローを設定を変えて実行する)あたりでエラーが発生して頭を悩ませる事態に陥るので気を付けてください。
遺伝子名を取得する 部分について
(2018.06.14 時点での内容です)
Galaxy ではデータライブラリからデータをインポートする方法を行おうとすると、データライブラリに "Training data" が見つからずインポートができないので、リンクからデータをインポートするか、ファイルをローカルに落としてからインポートしてください。
以上で From peaks to genes の補足説明を終わりにします。
ほかに分からないことがあればコメントか、Galaxy のセミナーに参加して私にお尋ねください。
リンクなど
- From peaks to genes の原文
(http://galaxyproject.github.io/training-material/topics/introduction/tutorials/galaxy-intro-peaks2genes/tutorial.html ) - From peaks to genes 日本語版
(https://github.com/A-Asai/training-material/blob/master/topics/introduction/tutorials/galaxy-intro-peaks2genes/tutorial_ja_notag.md ) - Galaxy
(https://usegalaxy.org/ ) - Galaxy Seminar
(https://pitagora.connpass.com/ )