なんかAgilentの解析に苦労したので書きます
使うソフト
RStudio使います。
データのあるディレクトリを作業ディレクトリに設定しておいてください。
タブにあるSession > Set Working Directory > Choose Directoryから設定できます。
下準備
データを扱う前には下準備が必要です。
読み込んで処理する対象ファイルを示してあげる必要があります。
↓こんな感じ
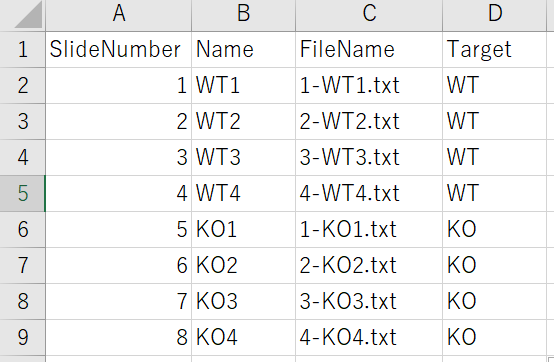
| 列名 | 内容 |
|---|---|
| SlideNumber | アレイデータの通し番号 |
| Name | 各アレイデータの通称。わかりやすい名前で。 |
| FileName | アレイデータの入ったファイルの名前 |
| Target | アレイデータの群分け。3群以上でも可 |
Excelとかで作ってTargets.txtとしてタブ区切りで保存しましょう。
(作業ディレクトリにおいてね)
パッケージのインスコ
データの読み込みとか正規化に使うLimmaパッケージをBiocManagerからインストールします。
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("limma")
インストールしたLimmaパッケージを読み込みます。
library(limma)
データの読み込み
生のtxtファイル使います。
↓Excelで開くとこんな感じのやつ
さっき作ったターゲットを指定するファイルを使ってデータを読み込みます。
targets <- readTargets("Targets.txt", row.names="Name")
RG <- read.maimages(targets, source="agilent",
green.only=TRUE,other.columns="gIsWellAboveBG")
データの正規化
バックグラウンドの補正と正規化をします。
boxplot(RG$E,ylim=c(0,300))
RGcor <- backgroundCorrect(RG, method="normexp")
boxplot(RGcor$E,ylim=c(0,300))
RGnorm <- normalizeBetweenArrays(RGcor, method="quantile")
※正規化
マイクロアレイではアレイデータごとに全体的なシグナル強度に差が出てしまう。
それだとアレイ間の比較が出来ないので、シグナル強度を合わせようっていう処理。
色んなメソッドの理解にはこのスライドが役立った。
https://www.slideshare.net/antiplastics/normalization-of-microarray
バックグラウンド補正のmethod="normexp"と正規化のmethod="quantile"から見るにRMA法に相当する処理をしてるっぽい。
遺伝子のフィルタリング
シグナル強度がバックグラウンドを下回るプローブ、Gene Symbol(Gapdhみたいな遺伝子の通称)がないプローブは除く。
Control <- RGnorm$genes$ControlType==1L
NoSymbol <- is.na(RGnorm$genes$GeneName)
IsExpr <- rowSums(RGnorm$other$gIsWellAboveBG > 0) >= 1
RGfilt <- RGnorm[!Control & !NoSymbol & IsExpr, ]
各アレイデータを群に割り当てる
最初に使ったTargets.txtのTarget列を使って、各アレイデータを群に振り分けます。
lev <- c("WT","KO")
f <- factor(targets$Target,levels=lev)
design <- model.matrix(~0+f)
colnames(design) <- lev
fit <- lmFit(RGfilt,design)
1行目は設定した群に応じて書き換えてください。
解析・プロット
contrast.matrix <- makeContrasts(KO-WT,levels=design)
fit2 <- contrasts.fit(fit, contrast.matrix)
plotMD(fit2)
1行目の「KO-WT」のところがキモです。
このステップまでに各群のシグナルがlog2変換されていますので、その差によってアレイ間の比較を行います。その引き算をここでやっているわけです。
多群の場合には「(A-B)-(C-D)」などとすることもできます。
実験デザインによって書き換えて使ってください。
出てくる散布図は横軸が発現量の平均、縦軸が発現変化量になってます。
特定の遺伝子をプロットするのはまた今度書きます。
補足
各群が2つ以上のデータを含んでいる場合は
fit2 <- eBayes(fit2)
とすることでベイズ統計的にp値などを求められるようです。
おわり
おわりです。
バイオインフォ的なことに触れるのは初めてだったので間違いがあるかもしれません。
間違いとかこうした方がいいよとかあったら、教えていただけると非常に非常にありがたいです。
今後少しずつパスウェイ解析なども書こうかと思います。