2019年4月25日追記:
一部の記述を修正。
目的
- GromacsのMD計算で得られたトラジェクトリファイルを適切に変換し、VMDやPyMOLなどで表示するための知識やメモ。
- 特に
gmx trjconvコマンド周辺を使った変換方法の説明。 - 水分子・溶媒イオンのトラジェクトリを必要としない場合、AmberToolsを使う方法の方が速い。
前提知識
-
Gromacsのトラジェクトリファイルは
trr形式とxtc形式がある。xtc形式はMDの設定(.mdpファイル内)に指定していなければ自動的に生成されない。trrは各粒子の座標+速度+時間情報を保存できるのに対し、xtcファイルは座標+時間情報のみ。 -
Gromacsのトラジェクトリファイルは原子の種類や残基の種類などの情報を含まない。反対に、これらの情報は
pdb、gro、tpr形式のファイルなら通常含んでいる。- したがって、VMDやPyMOLなどでタンパク質を見たい場合には、先に原子情報・残基情報を持っている
pdb、groを開いてタンパク質を構成する基本情報をロードし、その上で各タンパク質の座標の変化情報を持つトラジェクトリファイルをロードする必要がある。このとき、pdb、groを構成する粒子数と、トラジェクトリファイルを構成する粒子数が一致しないと、たいていエラーを吐かずにバグるので、常にこれらが一致しているかを確認する必要がある。
- したがって、VMDやPyMOLなどでタンパク質を見たい場合には、先に原子情報・残基情報を持っている
-
Gromacsコマンドは基本的に、拡張子でファイルタイプを判別する。中身を見て自動判定してくれることはしないはず。
-
Gromacsの周期境界条件(PBC)内でMDシミュレーションを行う場合、Gromacsトラジェクトリファイル内に保存される座標はそのPBCの外側に出ない。
- なので、何もしないまま
trrファイルをPyMOLで見ようとすると、こんな感じでタンパク質が途切れて見えることがほとんどである(内部処理的にはPBCでループしているため壊れていないが、人間には壊れているように見える)
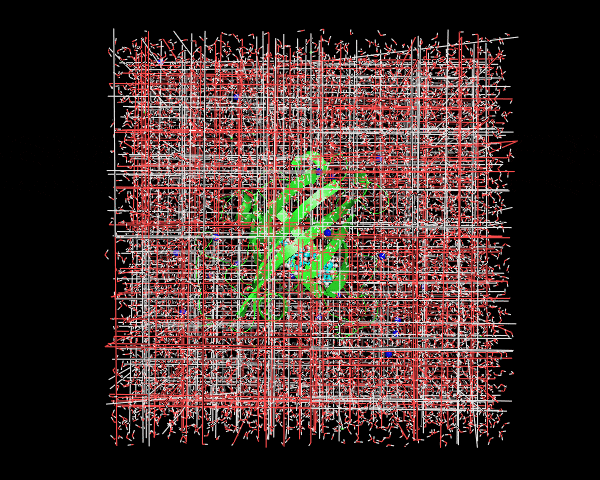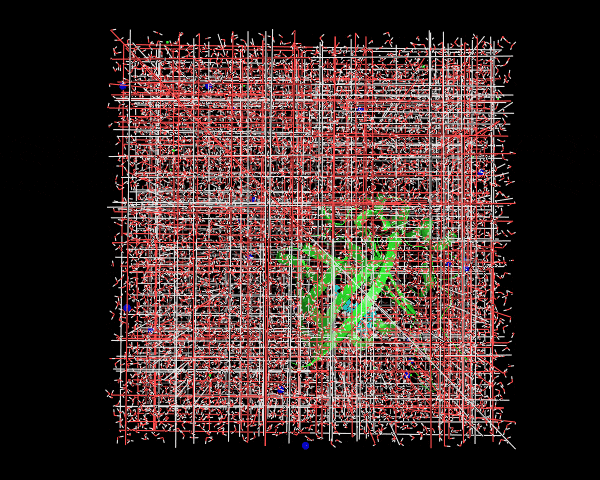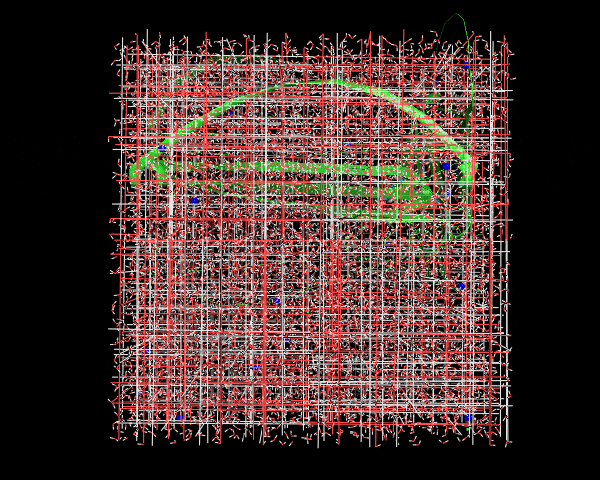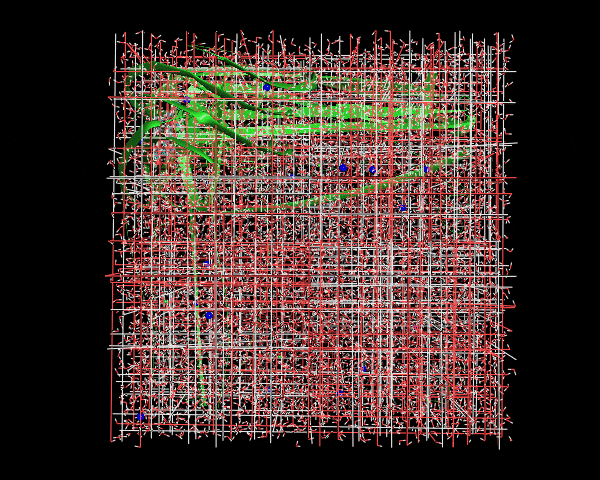
- なので、何もしないまま
-
以上から、Gromacsで計算されたMDトラジェクトリを人間の目で理解しやすくするためには、以下の操作を行うことが多い。
- タンパク質が途切れて表示されるのを修正する
- タンパク質の重心を平行移動操作によって固定&センタリング
- さらに、回転操作を用いてあるリファレンス構造から大きく回転しないようフィッティングさせる
- 場合によっては、水やイオンを見る必要はないのでこれらを適切に除去する
実際の操作
Gromacsで得られたtrrファイルをgmx trjconvで処理を施し、適切に見えやすくする。私が色々試した結果、トラジェクトリの変換操作は、
- PBCによる途切れを全体的に修正する(whole)
- さらにそのトラジェクトリをclusterごとに集める
- 続いてタンパク質(溶質)の重心の位置をセンタリングする
- さらに回転操作を行う
の順に変換を行うと、最も普遍的に問題なく処理を行うことができるようだ。これらの変換操作を一度に行うことはできない様子。gmx trjconvによるトラジェクトリ変換にはリファレンス構造を必要とし、gmx trjconvの-sに続くオプションで指定する。通常tpr形式が最も都合が良い。このとき、途切れた状態で保存されているtpr形式を-s入力に使うと表示がおかしくなるので十分に注意する。したがって、-sに入れるリファレンス構造は、トポロジーを作ってエネルギー最小化を始める前のtprファイルが最も都合が良い。
インデックスファイルの作成
gmx trjconvはトラジェクトリの変換を行ってくれるプログラムであり、引数-nにGROMACS形式のインデックスファイル(拡張子.ndx, 通常index.ndxとされることが多い)を指定することができる。インデックスファイルを指定しない場合でも最低限のグループ選択はできるような仕組みになっているが、インデックスファイルを作成しておくとグループ選択を自由に追加できるので、やっておいたほうが良い。
インデックスファイルを作るコマンドはgmx make_ndxである。さらに-fで何かしらの構造ファイルを与える。以下はここで用いるインデックスファイルの作成例である。タンパク質(PDB:1LKE)のMDシミュレーションで用いる座標ファイル1lke.groが存在したとして、これに対し
gmx make_ndx -f 1lke.gro
と入力する。すると続いて追加入力を求められる。
There are: 8527 Water residues
Analysing Protein...
Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups...
Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups...
0 System : 28284 atoms
1 Protein : 2605 atoms
2 Protein-H : 1338 atoms
3 C-alpha : 169 atoms
4 Backbone : 507 atoms
5 MainChain : 677 atoms
6 MainChain+Cb : 830 atoms
7 MainChain+H : 842 atoms
8 SideChain : 1763 atoms
9 SideChain-H : 661 atoms
10 Prot-Masses : 2605 atoms
11 non-Protein : 25679 atoms
12 Other : 62 atoms
13 DOG : 62 atoms
14 Na+ : 20 atoms
15 Cl- : 16 atoms
16 Ion : 36 atoms
17 DOG : 62 atoms
18 Na+ : 20 atoms
19 Cl- : 16 atoms
20 Water : 25581 atoms
21 SOL : 25581 atoms
22 non-Water : 2703 atoms
23 Water_and_ions : 25617 atoms
nr : group '!': not 'name' nr name 'splitch' nr Enter: list groups
'a': atom '&': and 'del' nr 'splitres' nr 'l': list residues
't': atom type '|': or 'keep' nr 'splitat' nr 'h': help
'r': residue 'res' nr 'chain' char
"name": group 'case': case sensitive 'q': save and quit
'ri': residue index
>
ここでは>を求められるたびに、以下のように追加入力を行う。qを押すと終了。
23 Water_and_ions : 25617 atoms
nr : group '!': not 'name' nr name 'splitch' nr Enter: list groups
'a': atom '&': and 'del' nr 'splitres' nr 'l': list residues
't': atom type '|': or 'keep' nr 'splitat' nr 'h': help
'r': residue 'res' nr 'chain' char
"name": group 'case': case sensitive 'q': save and quit
'ri': residue index
> !23
Copied index group 23 'Water_and_ions'
Complemented group: 2667 atoms
24 !Water_and_ions : 2667 atoms
> name 24 nowation
> q
ここではnowationという名前で「タンパク質+リガンドDOG(化合物名digoxigenin)」のグループを作成したいとする。これは「(系全体)-(水・イオン)」のグループと等価なので、この設定でグループを作成する。そこで追加入力の一度目は!23を入力する。これは23: Water_and_ionsに含まれる原子の補集合を表す!をつけることを意味する。これを指定すると上記のようなComplemented group 2667 atomsが24 !Water_and_ions : 2667 atomsとして出てくる(番号は系によって異なる)。この24番のグループを改名したい場合はname 24 nowationとする。最後にqを入力して終了する。
こうしてインデックスファイルindex.ndxの24番に「タンパク質+リガンドDOG(化合物名digoxigenin)」のグループが作成された。以降ではこうして作成したindex.ndxがディレクトリ../../top/index.ndxにあるものとして操作を行う。
1段階目の変換操作
まず1段階目の変換は
input=1lke
gmx trjconv -f ${input}.trr -o ${input}nj.trr -s ../minimize/min1.tpr -n ../../top/index.ndx -b 0 -e 20000 -skip 100 -pbc whole
ここで重要なのは、全体のトラジェクトリの途切れを修正する-pbc wholeの部分である。-bと-eのオプションではそれぞれ変換したいトラジェクトリの時間帯を指定できる。単位はpsなので、20000 ps = 20 ns。これは-tuオプションのデフォルトがpsになっているせい。詳細はgmx trjconv -hで確認できる。
これを実行すると、続けてこんな画面が出て追加入力を促される。
Command line:
gmx trjconv -f 1lke.trr -o 1lkenj.trr -s ../minimize/min1.tpr -n ../../top/index.ndx -b 0 -e 20000 -skip 100 -pbc whole
Will write trr: Trajectory in portable xdr format
Reading file ../minimize/min1.tpr, VERSION 2018 (single precision)
Reading file ../minimize/min1.tpr, VERSION 2018 (single precision)
Select group for output
Group 0 ( System) has 28284 elements
Group 1 ( Protein) has 2605 elements
Group 2 ( Protein-H) has 1338 elements
Group 3 ( C-alpha) has 169 elements
Group 4 ( Backbone) has 507 elements
Group 5 ( MainChain) has 677 elements
Group 6 ( MainChain+Cb) has 830 elements
Group 7 ( MainChain+H) has 842 elements
Group 8 ( SideChain) has 1763 elements
Group 9 ( SideChain-H) has 661 elements
Group 10 ( Prot-Masses) has 2605 elements
Group 11 ( non-Protein) has 25679 elements
Group 12 ( Other) has 62 elements
Group 13 ( DOG) has 62 elements
Group 14 ( Na+) has 20 elements
Group 15 ( Cl-) has 16 elements
Group 16 ( Ion) has 36 elements
Group 17 ( DOG) has 62 elements
Group 18 ( Na+) has 20 elements
Group 19 ( Cl-) has 16 elements
Group 20 ( Water) has 25581 elements
Group 21 ( SOL) has 25581 elements
Group 22 ( non-Water) has 2703 elements
Group 23 ( Water_and_ions) has 25617 elements
Group 24 ( nowation) has 2667 elements
Select a group:
ここの入力でアウトプット対象範囲を指定できる。番号または名前を入力するとその対象が出力される。
ここではまず水分子を含む全体のトラジェクトリのPBCによる分断を補正したいので、0またはSystemを入力する(どちらでも良い)。
先行入力したい場合には
input=1lke
gmx trjconv -f ${input}.trr -o ${input}nj.trr -s ../minimize/min1.tpr -n ../../top/index.ndx -b 0 -e 20000 -skip 100 -pbc whole <<EOF
0
EOF
というヒアドキュメント文法が使える。
上記のGroup選択のときにはndx形式のインデックスファイルなるもの(デフォルトはindex.ndx)を自分で作っておいて、gmx trjconv時にそのインデックスファイルを指定すれば良い。例えば水・イオン以外の全原子のGroupがほしいときには、このインデックスを作成しておいて、-nの引数で読み込ませて使う。index.ndx自体は単純なしくみになっているので簡単に後から手動で書き換えることもできると思う。この辺のGroup追加方法はマニュアルを見てください。
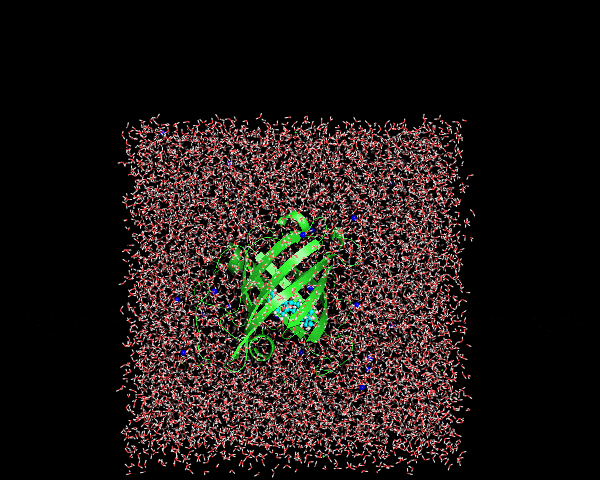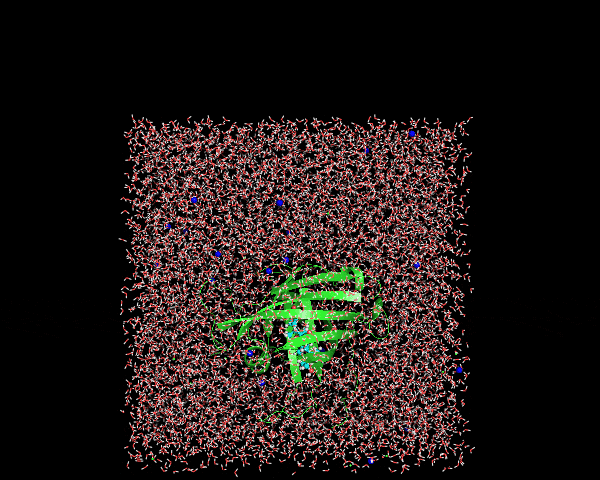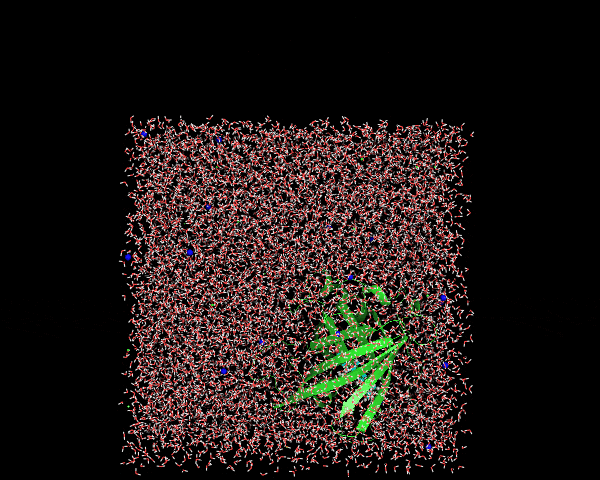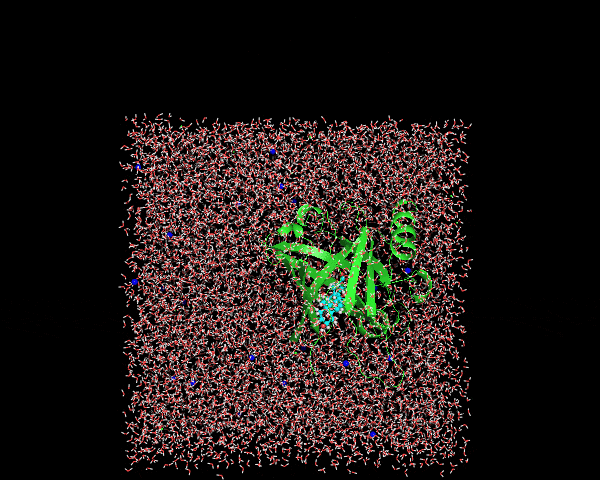
この時点で出てきた1lkenj.trrのトラジェクトリファイルをVMDやPyMOLなどで表示させてみると、こんな感じ
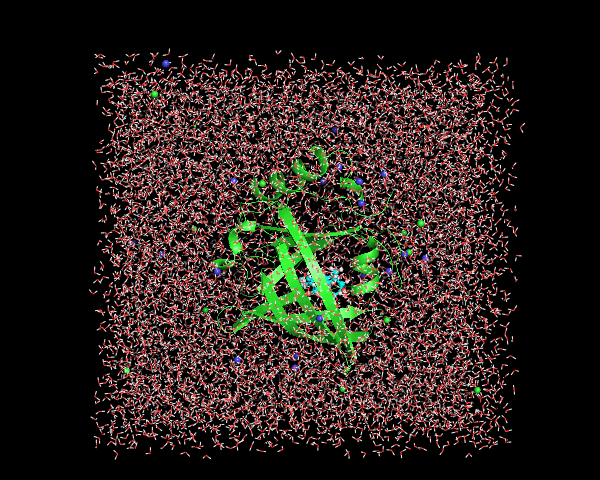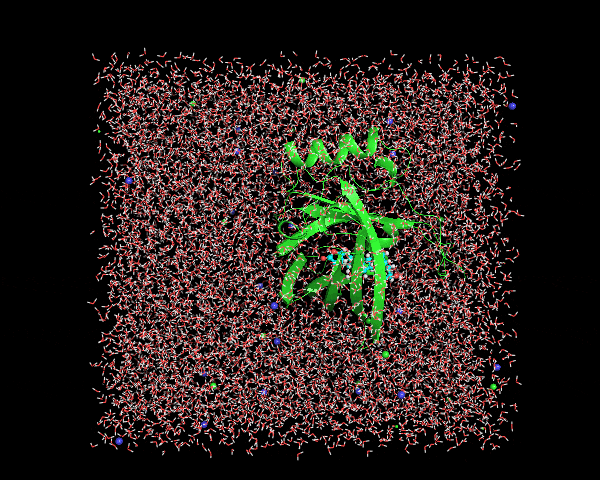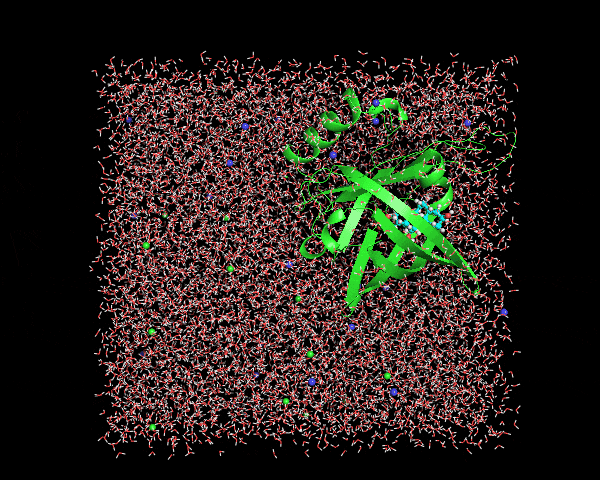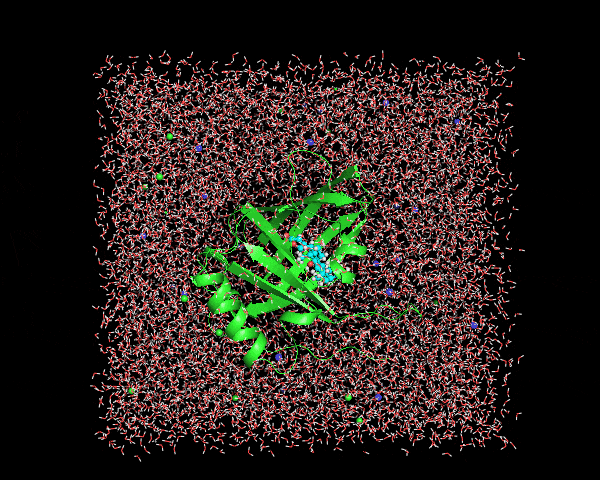
周期境界ボックスの周辺での水やタンパク質の途切れは修正されているけれど、一緒に結合している基質Digoxigenin(水色)とタンパク質(緑色)が分離しているフレームがいくつか存在している。この補正を2段階目で行う。
2段階目の操作
2段階目の操作は、1段階目で出てきた${input}nj.trrを入力として利用して
gmx trjconv -f ${input}nj.trr -o ${input}nj2.trr -s ../minimize/min1.tpr -n ../../top/index.ndx -pbc cluster
となる。すると、今度は1回目の追加入力が
Select group for clustering
Group 0 ( System) has 28284 elements
Group 1 ( Protein) has 2605 elements
Group 2 ( Protein-H) has 1338 elements
Group 3 ( C-alpha) has 169 elements
Group 4 ( Backbone) has 507 elements
Group 5 ( MainChain) has 677 elements
Group 6 ( MainChain+Cb) has 830 elements
Group 7 ( MainChain+H) has 842 elements
Group 8 ( SideChain) has 1763 elements
Group 9 ( SideChain-H) has 661 elements
Group 10 ( Prot-Masses) has 2605 elements
Group 11 ( non-Protein) has 25679 elements
Group 12 ( Other) has 62 elements
Group 13 ( DOG) has 62 elements
Group 14 ( Na+) has 20 elements
Group 15 ( Cl-) has 16 elements
Group 16 ( Ion) has 36 elements
Group 17 ( DOG) has 62 elements
Group 18 ( Na+) has 20 elements
Group 19 ( Cl-) has 16 elements
Group 20 ( Water) has 25581 elements
Group 21 ( SOL) has 25581 elements
Group 22 ( non-Water) has 2703 elements
Group 23 ( Water_and_ions) has 25617 elements
Group 24 ( nowation) has 2667 elements
Select a group:
となり、クラスタリングして表示したいグループの選択を要求される。ここで、1段階目ではうまくいっていなかった「タンパク質+Digoxigenin」のグループ、つまり24のnowationを同じ場所にクラスタリングするよう、指定してあげれば良い。
nowation(または24)と入力すると、続いてさらにSelect group for outputと表示され、出力してほしいグループを要求される。ここでは再び水・イオンを含んで全原子の表示を確認したいので、Systemまたは0を選択する。
この出来上がった1lkenj2.trrをPyMOLの上で表示するとこんな感じになっている。
先程はタンパク質とdigoxigeninが分離した状態のフレームが幾つか見られたが、ここでは常にこの2つが結合した状態で表示されているのがわかるだろう。周期境界の溶媒ボックスの外に飛び出してしまっているフレームの表示については次の段階で修正する。
3段階目の操作
3段階目は、タンパク質の重心が常に周期境界ボックスの中心にあるように移動させる変換を行う。
gmx trjconv -f ${input}nj2.trr -o ${input}nj3.trr -s ../minimize/min1.tpr -n ../../top/index.ndx -pbc mol -ur compact -center
例によってここではセンタリング対象とアウトプット対象を尋ねられるので、nowation,Systemと順に入力する。変換後はこんな感じ。
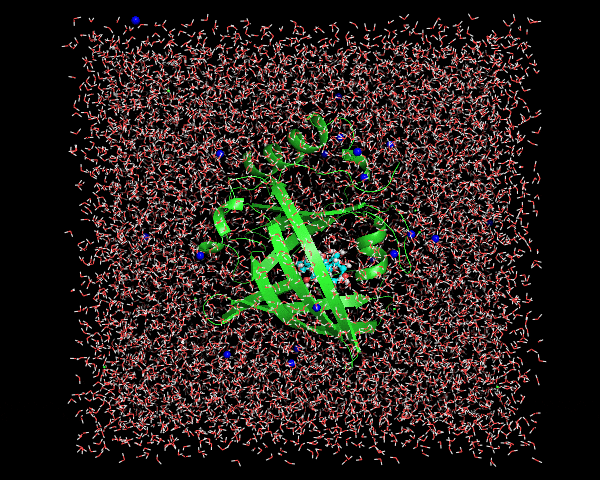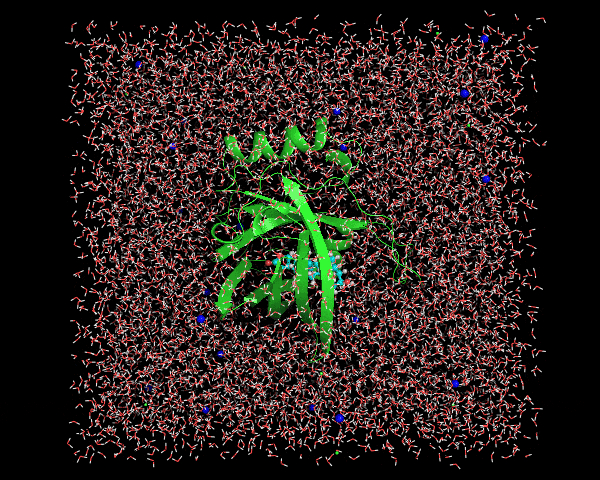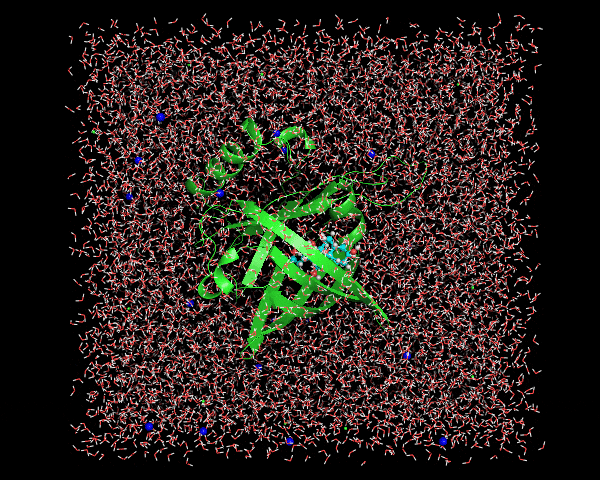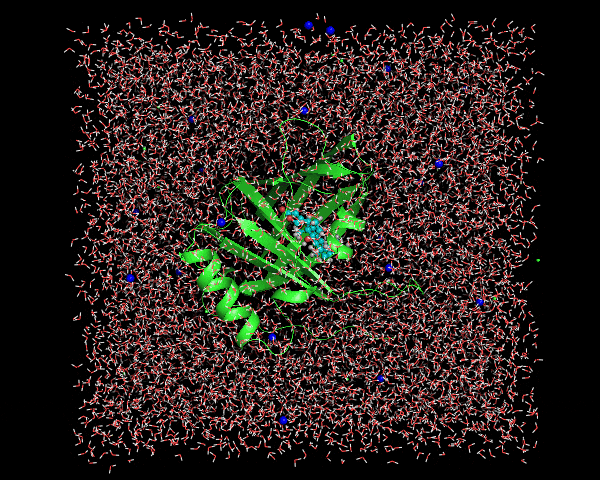
きれいにボックスの中にセンタリングされたタンパク質+digoxigeninが存在するのがわかる。
4段階目の操作
4段階目は、タンパク質の向きを一定に揃えることである。これはgmx trjconvのオプションの1つ-fit rot+transで揃えることができるのだが、これまでの-pbcオプションとタンパク質の向きを揃える-fit rot+transは同時に使えないらしいので、最後にこれを行う。
gmx trjconv -f ${input}nj3.trr -o ${input}nj4.trr -s ../minimize/min1.tpr -n ../../top/index.ndx -center -fit rot+trans
least squares fitの対象, centeringの対象、アウトプットの対象を問われるので、順にnowation, nowation, Systemの順で入力する。変換後はこんな感じ。
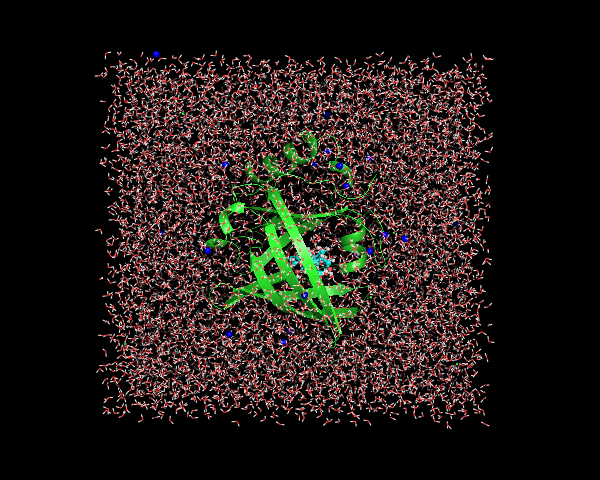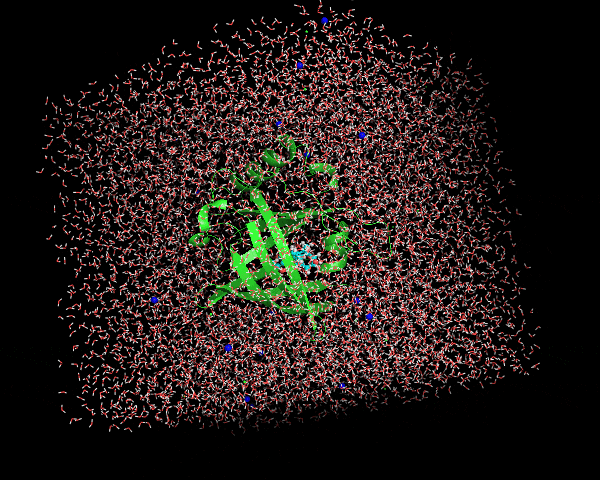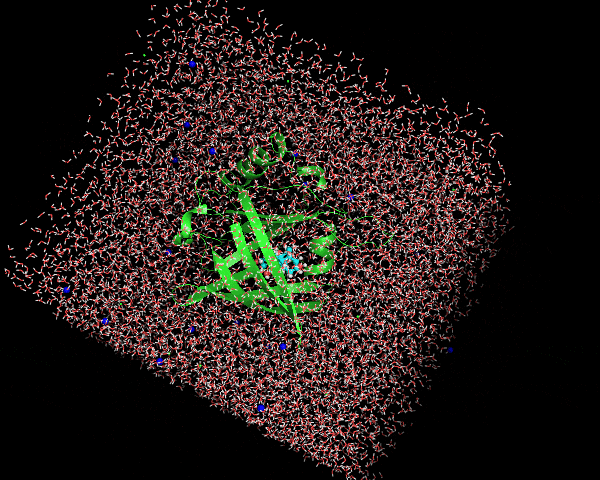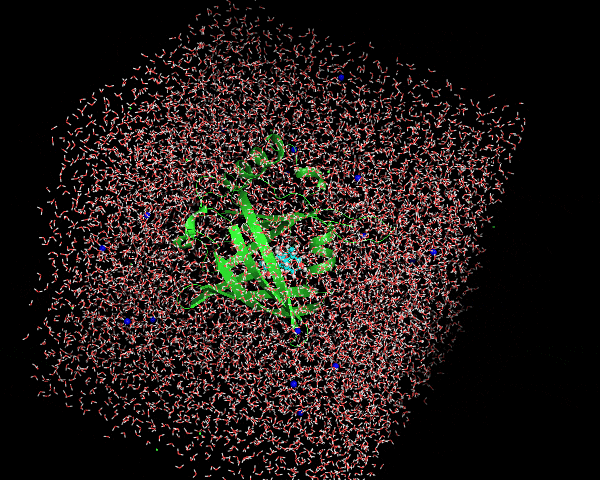
これで変換完了!!水分子を含むボックスがタンパク質の向きに合わせて回転されているのがわかります。今回のケースではN末端、C末端にやや長いループが存在することもあり、なんかタンパク質の重心がフレームごとに定まっていないように見えるが、ループ構造の少ないタンパク質ならばほとんど動かないように表示されるはず。
水・イオンのないトラジェクトリに変換&出力して表示させたい場合
より現実的な使い方として、シミュレーションの中に非常に多く存在する水分子やイオンを表示させる必要がないケースは多々ある。そんなときは、これまで見てきたアウトプット対象の指定をSystem、つまり全原子から、nowation、つまりタンパク質+digoxigeninだけにすれば、望み通りに水・イオンのないトラジェクトリファイルを作成することができる。
1段階目〜4段階目を水やイオンなしで順にまとめて処理するスクリプトはこちら。
# !/bin/sh
input=1lke
gmx trjconv -f ${input}.trr -o ${input}nj.trr -s ../minimize/min1.tpr -n ../../top/index.ndx -b 0 -e 100000 -skip 100 -pbc whole <<EOF || exit 1
nowation
EOF
gmx trjconv -f ${input}nj.trr -o ${input}nj2.trr -s ../minimize/min1.tpr -n ../../top/index.ndx -pbc cluster <<EOF || exit 1
nowation
nowation
EOF
gmx trjconv -f ${input}nj2.trr -o ${input}nj3.trr -s ../minimize/min1.tpr -n ../../top/index.ndx -pbc mol -ur compact -center <<EOF || exit 1
nowation
nowation
EOF
gmx trjconv -f ${input}nj3.trr -o ${input}nj4.trr -s ../minimize/min1.tpr -n ../../top/index.ndx -center -fit rot+trans <<EOF || exit 1
nowation
nowation
nowation
EOF
このような表示になる。

AmberToolsを使ってトラジェクトリを変換する方法
AmberToolsがインストールされており、かつ水・イオンのトラジェクトリを必要としない場合は、AmberToolsのcpptrajコマンドを使った方法の方が簡単かつ正確かつ処理も速いです。
AmberToolsにはcpptrajというモジュールが同梱されており、主としてトラジェクトリの処理や解析に用います。詳しくはAmberToolsのマニュアルのcpptrajの章を読んでください。
ここで、trajfix.inというファイルを用意して中に以下のように書き込んでおきます。
# trajinで処理させたいトラジェクトリ名を指定、その後の1 last 10は、1フレーム目から最後のフレーム目まで、10フレームおきに読み込むという意味
trajin 1lke.trr 1 last 10
# 残基番号で1から170に対応する残基を、同じBoundary box内に位置するようにする(ボックスの外に行ってしまっても途中でループさせない)
unwrap :1-170
# 残基番号1-170のCα原子の重心を原点上にセンタリングするよう、各フレームのトラジェクトリを平行移動させる(Gromacsの3段階目の操作に対応)
center :1-170@CA mass origin
autoimage
# 1フレーム目(first)をリファレンスとして、rmsが最小になるよう回転操作を行う(上記4段階目の操作に対応)
rms first out rmsd.dat @CA
# stripコマンドで、SOD, WAT……などの残基に対応するトラジェクトリを出力対象から外す(水・イオンのないトラジェクトリを作成するとき用)。今はコメントアウトしてある。
# strip :SOD,CLA,WAT,TIP3,Na+,Cl-
# 以上の処理を施したものを1lkenj4.trrという名前で出力する
trajout 1lkenj4.trr
go
このtrajfix.inファイルをMDトラジェクトリの1lke.trrと同じディレクトリに置き、cpptraj -p /path/to/leap.pdb -i trajfix.in と入力すれば(ここで/path/to/leap.pdbとは、対象のleap.pdbが置かれているディレクトリへのPATHを意味します)、上記の手法より圧倒的に早くかつ正確にトラジェクトリを変換してくれて、上の1lkenj4.trrと同じものが作成されます。
上の gmx trjconv と比べると、1発で変換でき、処理速度も5倍くらい速く、あまり深く考える必要がなく変換できます。
もし上記の方法でトラジェクトリが不審な動きをする(一部の原子が吹っ飛んでしまうなど)場合は、trajfix.inの1行目のtrajin 1lke.trr 1 last 10の10の数字を1など小さい値に変えることで解決することがあります。
これでも解決しない場合や、水やイオンを入れたままのトラジェクトリ変換を行いたい場合は、上記のgmx trjconvコマンドの方で(時間がかかりますが)解決することがあります。
(補足)水・イオンのないgroファイルの作成
上で挙げたように、トラジェクトリを載せる時用のpdbファイルやgroファイルを用意する必要があることもあるだろう。
このときのpdbファイルは単純に水やイオンのある行を消去すればよいだけである。
一方でgroファイルについては1つだけ気をつけることがある。
groファイルの仕様上、1行めはコメント、2行目に系の原子数が定義されているため、ここを修正しなければならない。例えば、溶媒和されているmin1.groファイルのコピーnowat.groを作成し、その中身を以下のように書き換える。
leap
2667
1ASP N 1 2.452 1.725 4.361
1ASP H1 2 2.355 1.752 4.347
1ASP H2 3 2.457 1.624 4.364
1ASP H3 4 2.506 1.758 4.283
(中略)
170DOG H26 2659 2.969 2.969 3.226
170DOG H27 2660 2.846 2.911 3.019
170DOG H28 2661 2.961 3.008 2.927
170DOG H29 2662 2.772 2.974 2.790
170DOG H30 2663 2.566 3.081 2.876
170DOG H31 2664 2.693 3.211 2.751
170DOG H32 2665 2.868 3.193 2.759
170DOG H33 2666 2.788 3.391 2.893
170DOG H34 2667 2.682 3.286 2.983
7.14920 7.29594 6.88980
こうすることで、2667原子からなるgroファイルに作り変えることが簡単に可能である。PyMOLやVMDでタンパク質を表示させるとき、必ずトラジェクトリに含まれる原子数と、ベースに載せるタンパク質の原子数は一致していなければならないことを覚えておく。
PyMOLを開く → 原子数が一致するpdbまたはgroファイルをロードして表示(オブジェクト名をnowatとする) → コマンドラインにload /path/to/1lkenj4.trr, nowat, state=1と入力して実行すればうまく表示されるはずである(コマンドラインを使うときは実行ディレクトリやパスに注意)。