BioPython
近年、データサイエンスの世界でよく使われるようになったプログラミング言語Pythonですが、C言語などと比べて簡易な文法や文字列操作の利便性から、生命科学の世界、特にバイオインフォマティクスの世界にもPythonは浸透してきました。1999年から開発が始まったBioPythonは生命科学における特有のデータ処理 (DNA・RNA・タンパク質配列や各種データベースとの連携など) を容易にしてくれます。
Pythonのインストール方法
macOSの場合
Pythonのインストール方法は色々ありますが、まずはHomebrewでインストールする方法を推奨します。Homebrewを一度インストールしていれば、引き続きターミナルから
brew install python@3.13
とすることで簡単に最新のPython 3.13.xをインストールすることができます(3はメジャーバージョン番号、13はマイナーバージョン番号、xはビルド(またはメンテナンス)バージョン番号と呼ぶ)。
Ubuntu 22.04の場合
デフォルトで入っているaptコマンドを使ってインストールすることができます。Ubuntuは使用上aptのようなシステム全体に影響を及ぼす(=同じマシンを使っている他のユーザーにも影響を与える)ようなコマンドの場合にはsudoをつけてから実行する必要があります。
sudo apt update
sudo apt -y upgrade
sudo apt -y python3 python3-pip
もしかしたら最初から何もしなくても使えるかもしれません。
BioPythonパッケージのインストール方法
Python3(3.10以上を推奨)が入っている状態で、Pythonのパッケージ管理システムpipを使うことでインストールすることができます。3.12以降のPythonを使っている場合は、何かしらの仮想環境を作成してからpipでインストールすることが強制されています。例えばkadaiEというディレクトリを作成し、その中に仮想環境を作成する場合は以下のようにします。
cd ~/Desktop/kadaiE
python3 -m venv .venv
. .venv/bin/activate
which python3
# /Users/Username/Desktop/kadaiE/.venv/bin/python3
python3 -m pip install biopython
Bio.Seqパッケージを使った塩基配列・アミノ酸配列操作
Pythonはターミナルから直接使うこともできますが、現実にはVSCodeをお使いのパソコンにインストールし、Pythonプラグインと, Jupyterプラグイン, Pylanceプラグインを併用するとかなり便利になります。
VSCodeでJupyterを動かすための準備としては、VS Code でPython,Jupyter を動かす の記事などを参照してください。# %%をつけておくと、VSCodeのJupyter上では# %%間に挟まれたブロックごとに実行することができます。
配列操作
塩基配列・アミノ酸配列についての文字列処理を行うには、from Bio.Seq import Seqを使います。これによりSeq()の関数を使うことができ、塩基配列またはアミノ酸配列を定義することができます。
Seqオブジェクトのメソッドとして、相補を返すcomplementや逆相補を返すreverse_complement, DNA→RNAへの転写(TをUに変える)transcribeやアミノ酸への翻訳translateが利用できます。
# 仮想環境の中でPythonを起動する。先にpip install biopythonをしておくこと。
$ python3
# 以降はPythonのインタラクティブシェル
Python 3.13.3 (main, Apr 8 2025, 13:54:08) [Clang 16.0.0 (clang-1600.0.26.6)] on darwin
Type "help", "copyright", "credits" or "license" for more information.
# BioPythonのインポート
>>> from Bio.Seq import Seq
# BioPythonのSeq()関数を使って変数my_seqを定義する
>>> my_seq = Seq("AGTACACTGGTA")
# 変数my_seqの中身を表示
>>> print(my_seq)
AGTACACTGGTA
# 変数my_seqの型を表示
>>> type(my_seq)
<class 'Bio.Seq.Seq'>
# 3塩基目から最後まで
>>> print(my_seq[2:])
TACACTGGTA
# 先頭から4塩基目まで
>>> print(my_seq[:4])
AGTA
# 逆順に返す
>>> print(my_seq[::-1])
ATGGTCACATGA
# my_seqの相補鎖
>>> print(my_seq.complement())
TCATGTGACCAT
# my_seqの逆相補鎖
>>> print(my_seq.reverse_complement())
TACCAGTGTACT
#転写
>>> print(my_seq.transcribe())
AGUACACUGGUA
#翻訳
>>> print(my_seq.translate())
STLV
適当なところでCtrl-Dを押すとPythonのインタラクティブシェルを終了できます。
Bio.SeqIOを使ってアミノ酸配列データを取得する
PythonとBioPythonの機能だけを使ってあるFASTA形式のファイルをダウンロードし、そのアミノ酸配列を取得する方法を示します。例として、タマネギの催涙因子を生成するタンパク質lachrymatory factor synthase (LFS)のUniprot ID:P59082をダウンロードしてからそのアミノ酸配列の一部領域を表示するスクリプトを示します。
https://www.uniprot.org/uniprot/P59082 がUniprotにおけるLFSのエントリのページです。このページのSequence見出しのところから、FASTA(読み方はファストエー、ファスタではない)形式でのアミノ酸配列データのダウンロードが可能です。https://www.uniprot.org/uniprot/P59082.fasta が配列データへのhttpsアドレスです。
>sp|P59082|LFS_ALLCE Lachrymatory-factor synthase OS=Allium cepa OX=4679 GN=LFS PE=1 SV=1
MELNPGAPAVVADSANGARKWSGKVHALLPNTKPEQAWTLLKDFINLHKVMPSLSVCELV
EGEANVVGCVRYVKGIMHPIEEEFWAKEKLVALDNKNMSYSYIFTECFTGYEDYTATMQI
VEGPEHKGSRFDWSFQCKYIEGMTESAFTEILQHWATEIGQKIEEVCSA
まずはFASTA形式のsequence recordの書式を理解しておきます。テンプレートは
>{name} {description}?
{sequence}
となっており、
>sp|P59082|LFS_ALLCE Lachrymatory-factor synthase OS=Allium cepa OX=4679 GN=LFS PE=1 SV=1
MELNPGAPAVVADSANGARKWSGKVHALLPNTKPEQAWTLLKDFINLHKVMPSLSVCELV
EGEANVVGCVRYVKGIMHPIEEEFWAKEKLVALDNKNMSYSYIFTECFTGYEDYTATMQI
VEGPEHKGSRFDWSFQCKYIEGMTESAFTEILQHWATEIGQKIEEVCSA
という例においては、nameがsp|P59082|LFS_ALLCEに相当し、descriptionがLachrymatory-factor synthase OS=Allium cepa OX=4679 GN=LFS PE=1 SV=1、sequenceがMELNPG...のアミノ酸配列部分です。
BioPythonのSeqIOモジュールの関数の1つであるSeqIO.parse()関数を用いると、ある1つのFASTAフォーマットのデータに含まれているname, description, sequenceをそれぞれ適切に取り込むことができます。その後、SeqIOのattributeのid, description, seqを使うことで各データを読み出すことができます。
以上を踏まえて、P59082.fasta内の全record(※recordはP59082.fastaというファイルには1つだけです)に含まれる情報(name,description, sequence)をすべて表示したい場合は、Pythonで以下のように記述します。
# URLを開くためのurllib.requestライブラリを使えるようにimportする
import urllib.request
from Bio import SeqIO
# URL:https://www.uniprot.org/uniprot/P59082.fasta の内容を "P59082.fasta"という名前で保存する
urllib.request.urlretrieve(
"https://www.uniprot.org/uniprot/P59082.fasta", "P59082.fasta"
)
fastafile = "P59082.fasta"
with open(fastafile, "r") as handle:
for record in SeqIO.parse(handle, "fasta"):
# 変数id_partにrecordのname(ID)を代入する
id_part = record.id
# 変数desc_partにrecordのdescriptionを代入する
desc_part = record.description
# 変数seqにrecordのseq(sequence)を代入する
seq = record.seq
# print()で表示
print("id:", id_part)
print("desc:", desc_part)
print("seq:", seq)
print("Residues 1-10 of the sequence are " + seq[0:10])
実行結果はこのようになります。
id: sp|P59082|LFS_ALLCE
desc: sp|P59082|LFS_ALLCE Lachrymatory-factor synthase OS=Allium cepa OX=4679 GN=LFS PE=1 SV=1
seq: MELNPGAPAVVADSANGARKWSGKVHALLPNTKPEQAWTLLKDFINLHKVMPSLSVCELVEGEANVVGCVRYVKGIMHPIEEEFWAKEKLVALDNKNMSYSYIFTECFTGYEDYTATMQIVEGPEHKGSRFDWSFQCKYIEGMTESAFTEILQHWATEIGQKIEEVCSA
Residues 1-10 of the sequence are MELNPGAPAV
Pythonの文字列操作でおなじみのスライス機能[0:10]を入れると、指定範囲のアミノ酸を切り出すことができます。Pythonは0始まりであることに気をつけてください。
BioPythonのSeq, SeqIOライブラリの具体的な使い方、APIの定義はSeqライブラリの公式WikiやSeqIOライブラリの公式Wikiを参照してください。
gzip圧縮されたFASTA形式ファイルから所望のデータを取り出す
FASTAフォーマットのファイルは1つのファイルに複数のレコードを持たせることもできます(というかこっちの方が普通)。例としてPDBの全構造情報のアミノ酸配列をFASTA形式で配信しているファイルpdb_seqres.txt.gz(32MB)の中を調べてみます。以下は、作業ディレクトリの中からターミナルから入力してください。
$で始まる行はターミナル上で実行するコマンドを示しています。>>>で始まる行はPythonのインタラクティブシェル上で実行するコマンドを示しています。
# インターネットからpdb_seqres.txt.gzを取得
$ curl https://files.wwpdb.org/pub/pdb/derived_data/pdb_seqres.txt.gz -o pdb_seqres.txt.gz
# gzipファイルを解凍(-kをつけることで元ファイルを保持する)
$ gunzip -k pdb_seqres.txt.gz
# 最初の数行を表示するheadコマンドでpdb_seqres.txtの中身を表示する
$ head pdb_seqres.txt
pdb_seqres.txt.gzはgunzipコマンドで解凍すると120MB以上の大きなファイルになります。headコマンドで中身を見てみますと
>100d_A mol:na length:10 DNA/RNA (5'-R(*CP*)-D(*CP*GP*GP*CP*GP*CP*CP*GP*)-R(*G)-3')
CCGGCGCCGG
>100d_B mol:na length:10 DNA/RNA (5'-R(*CP*)-D(*CP*GP*GP*CP*GP*CP*CP*GP*)-R(*G)-3')
CCGGCGCCGG
>101d_A mol:na length:12 DNA (5'-D(*CP*GP*CP*GP*AP*AP*TP*TP*(CBR)P*GP*CP*G)-3')
CGCGAATTCGCG
>101d_B mol:na length:12 DNA (5'-D(*CP*GP*CP*GP*AP*AP*TP*TP*(CBR)P*GP*CP*G)-3')
CGCGAATTCGCG
>101m_A mol:protein length:154 MYOGLOBIN
MVLSEGEWQLVLHVWAKVEADVAGHGQDILIRLFKSHPETLEKFDRVKHLKTEAEMKASEDLKKHGVTVLTALGAILKKKGHHEAELKPLAQSHATKHKIPIKYLEFISEAIIHVLHSRHPGNFGADAQGAMNKALELFRKDIAAKYKELGYQG
...
という中身が見られます。
次のコードでは、pdb_seqres.txt.gz内の全レコードを表示してくれます(が、非常に長いのでもし実行したのならCtrl-Cキーを押して実行を中止させましょう)
# gzipファイルのサポートライブラリ
import gzip
# 以下の2行がないとSSL: CERTIFICATE_VERIFY_FAILED エラーが発生し、files.wwpdb.org/pub/pdb/derived_data/pdb_seqres.txt.gz からファイルがダウンロードできない(2021年4月21日現在)
# おそらくpdbjのウェブサイト側がSSL認証を誤っているため、誤っていてもファイルをダウンロード可能にするためのSSL設定を追加する。
# wwpdb( https://files.wwpdb.org/pub/pdb/derived_data/pdb_seqres.txt.gz )からpdb_seqres.txt.gzをダウンロードすることもできるが、
# 日本からのダウンロードは非常に時間がかかるので、可能ならば日本のミラーサイトからダウンロードしたい。
import ssl
# URLを開くためのurllib.requestライブラリを使えるようにimportする
import urllib.request
from Bio import SeqIO
ssl._create_default_https_context = ssl._create_unverified_context
# 実行ディレクトリ上にpdb_seqres.txt.gzがなければ、インターネットからそれをダウンロードしてくる
if not os.path.exists("pdb_seqres.txt.gz"):
urllib.request.urlretrieve(
"https://files.wwpdb.org/pub/pdb/derived_data/pdb_seqres.txt.gz",
"pdb_seqres.txt.gz",
)
fastafile = "pdb_seqres.txt.gz"
# gzipファイルを開く場合、openの代わりにgzip.open()を使う。
# mode="rt"を指定しないとテキストデータとして読み込めないので注意。
with gzip.open(fastafile, mode="rt") as handle:
# pdb_seqres.txtの中の配列データを、1つの配列データごとにrecordという変数の中に入れる
for record in SeqIO.parse(handle, "fasta"):
id_part = record.id
desc_part = record.description
seq = record.seq
print("id:", id_part)
print("desc:", desc_part)
print("seq:", seq)
ここで、nameが102m_Aに合致するもののアミノ酸配列を表示したいのであれば、if文を使って以下のように書くと良いでしょう。
import gzip
import os
import ssl
import urllib.request
from Bio import SeqIO
ssl._create_default_https_context = ssl._create_unverified_context
# 実行ディレクトリ上にpdb_seqres.txt.gzがなければ、インターネットからそれをダウンロードしてくる
if not os.path.exists("pdb_seqres.txt.gz"):
urllib.request.urlretrieve(
"https://files.wwpdb.org/pub/pdb/derived_data/pdb_seqres.txt.gz",
"pdb_seqres.txt.gz",
)
fastafile = "pdb_seqres.txt.gz"
with gzip.open(fastafile, "rt") as handle:
for record in SeqIO.parse(handle, "fasta"):
# if文を使うことで、recordのidが"102m_A"に完全に一致するときのみ、以下を実行する
if record.id == "102m_A":
print(f"The Sequence is {record.seq}")
print(f"The length is {len(record.seq)}")
print(f"The description is {record.description}")
結果は以下のようになります。
The Sequence is MVLSEGEWQLVLHVWAKVEADVAGHGQDILIRLFKSHPETLEKFDRFKHLKTEAEMKASEDLKKAGVTVLTALGAILKKKGHHEAELKPLAQSHATKHKIPIKYLEFISEAIIHVLHSRHPGNFGADAQGAMNKALELFRKDIAAKYKELGYQG
The length is 154
The description is 102m_A mol:protein length:154 MYOGLOBIN
この結果は上述の102m_Aについてのアミノ酸配列
>102m_A mol:protein length:154 MYOGLOBIN
MVLSEGEWQLVLHVWAKVEADVAGHGQDILIRLFKSHPETLEKFDRFKHLKTEAEMKASEDLKKAGVTVLTALGAILKKKGHHEAELKPLAQSHATKHKIPIKYLEFISEAIIHVLHSRHPGNFGADAQGAMNKALELFRKDIAAKYKELGYQG
と一致しています。
このように、import gzipをしてからgzip.open(fastafile, "rt")をすることで、gzip圧縮ファイル状態になっているpdb_seqres.txt.gzから展開しないまま所望のデータを抜き出すことが可能です。この機能をうまく使っていきましょう。
ちなみに以下のようにすると、インプットとして入れるfastaファイルがgzip圧縮してある・していないの両方の場合に対応できます。
# gzip圧縮・非圧縮両対応のコード
import gzip
import urllib.request
from functools import partial
from mimetypes import guess_type
from Bio import SeqIO
# fastafile = "P59082.fasta.gz"
fastafile = "P59082.fasta"
# 以下の2行を加えることで、gzipしてある/してないの両方に対応できるようになる
encoding = guess_type(fastafile)[1] # uses file extension
_open = (
partial(gzip.open, mode="rt") if encoding == "gzip" else open
) # _openという関数をif文によって分岐して定義する
with _open(fastafile) as handle:
for record in SeqIO.parse(handle, "fasta"):
print("id:", record.id)
print("desc:", record.description)
print("seq:", record.seq)
テキストファイルをgzip圧縮・展開したい場合は、ターミナル上で以下のコマンドを入力します。
# P59082.fastaを圧縮してP59082.fasta.gzに変換する。-kをつけると元のファイルを維持する(keep)。
$ gzip P59082.fasta -k
# P59082.fastaを展開してP59082.fastaに変換する。-kをつけると元のファイルを維持する(keep)。
$ gunzip P59082.fasta.gz -k
Bio.PDBパッケージを使ったタンパク質構造情報の操作
Bio.PDBパッケージとは
Bio.PDBパッケージは生体分子、主にタンパク質の立体構造情報を処理するのに使われるパッケージです。Protein Data Bankで規格化され提供されているPDBフォーマット、mmCIFフォーマットの構造情報に対して効果を発揮します。また、近年ではさらにファイルサイズが軽量化されているmmtfフォーマットについても対応しています。
Bio.PDBパッケージはPDB, mmCIFファイルのファイルI/Oに用いるパーサーや、SMCRAデータ構造のクラス、自前サーバーにPDBデータのローカルコピーを作るためのツール……などなどを含んでいます。Bio.PDBパッケージ以下のすべてのサブパッケージ、サブモジュールは https://biopython.org/docs/latest/api/Bio.PDB.html から確認できます。
構造情報のダウンロード
Bio.PDBパッケージのモジュールであるPDBListを利用するために、from Bio.PDB.PDBList import PDBListを宣言し、PDBListクラスのメソッドのretrieve_pdb_file()を実行することでRCSB PDBのウェブサイト(※デフォルトはhttps://files.wwpdb.org)から指定したPDB IDの構造ファイルを直接ダウンロードすることができます(※当然インターネット接続が必要です)。
from Bio.PDB.PDBList import PDBList
# PDBListクラスのインスタンスを生成し、変数pdblに代入
pdbl = PDBList()
# ダウンロードしたいPDB IDのリストを入力
pdb_ids = ["1ALK", "1EW2", "5GTE"]
# retrieve_pdb_fileメソッドを使ってダウンロードする場合
for pdb_id in pdb_ids:
# pdirにはダウンロード先のディレクトリ名を、file_formatにはファイルフォーマットを指定する
# overwrite=trueにすると、すでに存在していても再ダウンロードする(デフォルトはFalse)。
pdbl.retrieve_pdb_file(pdb_id, pdir="pdb_files/", file_format="pdb", overwrite=True)
# またはdownload_pdb_filesメソッドを使うとリストを入力としてダウンロードすることができる
pdbl.download_pdb_files(pdb_ids, pdir="pdb_files/", file_format="pdb", overwrite=True)
詳細な文法は公式マニュアルを読みましょう。file_formatを指定しない場合、デフォルトではmmCIF形式でダウンロードします。ここではあえてfile_format="pdb"として、レガシーなPDBフォーマットをダウンロードしています。いずれの場合も、仕様としてまずgzip状態のままダウンロードし、その後自動的に解凍してくれます。ただ、PDBフォーマットの場合のみ、1alk.pdbのような命名法ではなく、pdb1alk.entのような形式になっているため、取り扱いには気をつけてください。これはProtein Data Bankのデータサーバー内にて保存されている正式なファイル名です。
file_formatはmmCif(デフォルト、PDBx/mmCif), pdb, xml(PMDML/XML), mmtf, bundle(巨大構造用PDBフォーマット、今はあまり使われない)を選択することができます。
巨大な複合体形成している場合など、いくつかのPDBエントリでは、表現能力の限界のために伝統的なPDBフォーマットが利用不可能になっていることがあります。このような場合はmmCIF形式でダウンロードしましょう。
PDBのSMCRAデータ構造
ダウンロードしてきた生体分子の立体構造情報についての取り扱いを説明するためには、BioPythonがどのように立体構造情報をパースして専用の データ構造 (data structure) に落とし込んでいるかを理解しておく必要があります。
BioPythonはまずタンパク質構造情報を Structure-Model-Chain-Residue-Atom frameworkのSMCRAデータ構造に変換します。(参考論文: PDB file parser and structure class implemented in Python)
この機能を利用するためには、まずfrom Bio.PDB.MMCIFParser import FastMMCIFParserを宣言し、FastMMCIFParserクラスのメソッドget_structure()を利用します。ちなみにFastMMCIFParserと同等の機能を持つMMCIFParserクラスも存在していますが、現在はFastMMCIFParserを使う方が良いでしょう。参考:https://biopython.org/docs/latest/api/Bio.PDB.MMCIFParser.html
以下では3つのやり方でPDB ID: 1ALKのmmCIFフォーマットの構造情報をダウンロードし、get_structure()でSMCRAデータ構造に変換してから変数strucに代入する方法を示します。
import gzip
import urllib.request
from mimetypes import guess_type
from Bio.PDB.MMCIFParser import FastMMCIFParser
from Bio.PDB.PDBList import PDBList
"""
やり方その1
"""
pdb_id = "1ALK"
# PDBListクラスのインスタンスを生成し、変数pdblに代入
pdbl = PDBList()
# 1ALKの構造データをmmCIF形式でpdb_files/ディレクトリにダウンロード(./pdb_files/1alk.cif)
pdbl.retrieve_pdb_file(pdb_id, pdir="pdb_files/", file_format="mmCif", overwrite=True)
# クラスMMCIFParserのインスタンスを生成する
pdb_parser = FastMMCIFParser()
# 変数strucに"./pdb_files/1alk.cif"ファイルの中身をパースして取り込む
struc = pdb_parser.get_structure("1ALK", "pdb_files/1alk.cif")
# strucの中身を表示する
print(struc)
"""
やり方その2, gzipファイルを使う場合
"""
pdb_parser = FastMMCIFParser()
# urllib.request.urlretrieve("https://files.rcsb.org/download/1EN2.cif.gz", "1EN2.cif.gz")の返り値は
# tuple型で('1EN2.cif.gz', <http.client.HTTPMessage object at 0x1589790a0>) のように仕様として決まっているので
# ciffile = '1EN2.cif.gz', httpobject = <http.client....> という変数にそれぞれ代入する
ciffile, httpobject = urllib.request.urlretrieve(
"https://files.rcsb.org/download/1ALK.cif.gz", "1ALK.cif.gz"
)
# gzipファイルの場合はwith gzip.open()とmode="rt"を使い,
# ファイルハンドルをpdb_parser.get_structure()関数の第2引数にする
with gzip.open("1ALK.cif.gz", mode="rt") as handle:
struc = pdb_parser.get_structure("1ALK", handle)
print(struc)
"""
やり方その3, openをgzip圧縮・非圧縮両対応にする場合
"""
# 上記のgzip両対応できるようになる_openを関数定義した場合の書き方
def _open(file):
encoding = guess_type(file)[1] # uses file extension
if encoding == "gzip":
return gzip.open(file, mode="rt")
else:
return open(file)
pdb_parser = FastMMCIFParser()
ciffile, httpobject = urllib.request.urlretrieve(
"https://files.rcsb.org/download/1ALK.cif.gz", "1ALK.cif.gz"
)
# ciffile = "./pdb_files/1alk.cif" # どちらでも可能
with _open(ciffile) as handle:
struc = pdb_parser.get_structure(
"1ALK", handle
) # "(strucuture_id, filename OR text mode file handle)"
print(struc)
実行すると、いくつかのWARNINGメッセージが現れていますが、最後のprint(struc)の部分で、ダウンロードしてきた1ALK.cif.gzファイルの中身をパースできている結果<Structure id=1ALK>が表示されます。(ちなみにこのWARNINGメッセージはpdb_parser = FastMMCIFParser(QUIET=True)とすると表示されなくなります)
Downloading PDB structure '1alk'...
<Structure id=1ALK>
<Structure id=1ALK>
<Structure id=1ALK>
/Users/YoshitakaM/Desktop/kadaiE/.venv/lib/python3.13/site-packages/Bio/PDB/StructureBuilder.py:100: PDBConstructionWarning: WARNING: Chain A is discontinuous at line 6608.
warnings.warn(
/Users/YoshitakaM/Desktop/kadaiE/.venv/lib/python3.13/site-packages/Bio/PDB/StructureBuilder.py:100: PDBConstructionWarning: WARNING: Chain B is discontinuous at line 6616.
warnings.warn(
/Users/YoshitakaM/Desktop/kadaiE/.venv/lib/python3.13/site-packages/Bio/PDB/StructureBuilder.py:100: PDBConstructionWarning: WARNING: Chain A is discontinuous at line 6624.
warnings.warn(
/Users/YoshitakaM/Desktop/kadaiE/.venv/lib/python3.13/site-packages/Bio/PDB/StructureBuilder.py:100: PDBConstructionWarning: WARNING: Chain B is discontinuous at line 6627.
warnings.warn(
/Users/YoshitakaM/Desktop/kadaiE/.venv/lib/python3.13/site-packages/Bio/PDB/StructureBuilder.py:100: PDBConstructionWarning: WARNING: Chain A is discontinuous at line 6608.
warnings.warn(
上の例では、PDBフォーマットの1ALK.cifについてpdb_parser.get_structure()関数で立体構造情報を読み込みます。このとき、各データは以下の図のように読み込まれます。
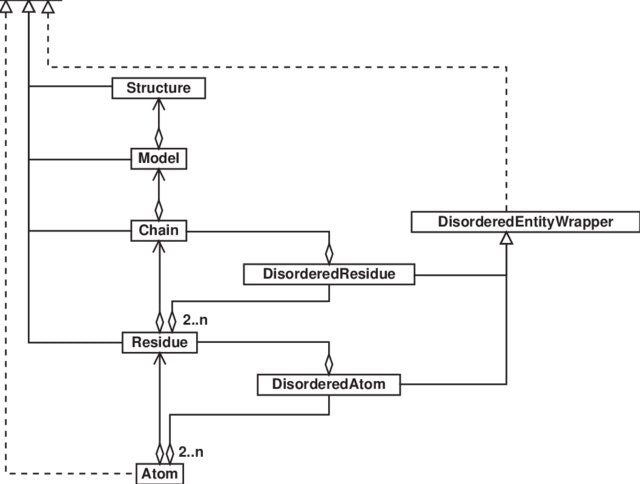
基本的にはStructureクラスの下にModel, その下にChain, Residue, Atomが続くデータクラス構造です。また、DisorderedAtom/Residueの取り扱いをまとめるDisorderedEntityWrapperのクラスもあり、StructureクラスとまとめてEntityクラスを形成します。ただ、Disorderedについては存在しない場合も多いので、ちょっと重要性は落ちます。
Disorderedについての取り扱いを無視すれば、以下のようなクラス構造になります。
PDB.Parser
|
|---structure
| |
| |--Model
| |
| |--Chain
| |
| |--Residue
| |
| |--Atom
|---structure
: :
: :
つまりSMCRAデータ構造とはBioPythonの中で定義されるタンパク構造情報についてのデータアーキテクチャのことで、PDBで配信されている構造情報をうまく落とし込むことができる作りになっています。言葉で表現し直すと
-
Structureオブジェクトは1つまたは複数のModelオブジェクトで構成される -
Modelオブジェクトは1つまたは複数のChainオブジェクトで構成される -
Chainオブジェクトは1つまたは複数のResidueオブジェクトで構成される -
Residueオブジェクトは1つまたは複数のAtomオブジェクトで構成される
という仕組みになっています。
Atom, Residue, Chainについてはタンパク質・DNAの構造式をイメージするとわかりやすいでしょう。Atomの集合体が1つのアミノ酸残基(Residue)を作り、複数のResidueがペプチド結合でつながっていった結果、1つのポリペプチド鎖(Chain)を形成するという化学・生物学的な知識と合致するでしょう。このポリペプチド鎖が決まった立体構造を取るように折りたたまれた状態が生化学で言うところの 三次構造(Tertiary Structure) です。そして複数の三次構造が合体した状態(タンパク質でいうとホモ二量体やヘテロ二量体、タンパク質-DNA複合体など)のことを 四次構造 (quaternary structure) と呼びます。SMCRAデータ構造内では、この四次構造がModelに該当すると思ってください。
最後に"Structureオブジェクトは1つまたは複数のModelオブジェクトで構成される"の部分だけは直感的に理解しにくいところがあると思うので、説明を補足します。PDBで配信されている構造情報の多くは 結晶構造 (Crystal Structure) であり、静止した1つの状態で提供されています。しかしながら、タンパク質を含めあらゆる分子は熱運動をしており、分子を構成する各原子間の結合長や角度、二面角は常時変動しています。そして、PDBで配信されている構造情報のエントリの中にはこうした分子の「動き」を収録しているものも存在します。特に、核磁気共鳴法(NMR)という構造決定法を用いて決定された構造情報にはこの「動き」が含まれていることが多いです。
例としてPDB: 2C0SのPDBエントリを見てみます。PyMOLを使って構造情報を開いてみますと64残基から構成されるタンパク質が画面に表示されますが、ここで右下のStateに注目すると、25のStateが存在することがわかります。

この25がModelの数であり、SMCRAデータ構造に則って言えば、"PDB: 2C0Sにおいて、1つのStructureオブジェクトには25のModelオブジェクトがある"ということを表しています。また、PDB: 2C0Sについての構造情報のPDBフォーマットファイルを任意のテキストエディタを使って開いてみますと
MODEL 1
ATOM 1 N MET A 1 9.917 -5.433 3.492 1.00 0.00 N
ATOM 2 CA MET A 1 11.334 -5.529 3.105 1.00 0.00 C
ATOM 3 C MET A 1 11.496 -5.854 1.630 1.00 0.00 C
...
ATOM 1130 HD1 HIS A 64 17.948 -3.271 5.475 1.00 0.00 H
ATOM 1131 HD2 HIS A 64 18.090 -7.386 5.888 1.00 0.00 H
ATOM 1132 HE1 HIS A 64 16.565 -4.216 3.567 1.00 0.00 H
TER 1133 HIS A 64
ENDMDL
MODEL 2
ATOM 1 N MET A 1 12.599 -6.593 -1.354 1.00 0.00 N
ATOM 2 CA MET A 1 13.144 -7.195 -2.558 1.00 0.00 C
ATOM 3 C MET A 1 12.800 -6.352 -3.786 1.00 0.00 C
ATOM 4 O MET A 1 13.693 -5.848 -4.460 1.00 0.00 O
...
ATOM 1130 HD1 HIS A 64 2.264 -11.207 5.125 1.00 0.00 H
ATOM 1131 HD2 HIS A 64 5.458 -10.976 7.761 1.00 0.00 H
ATOM 1132 HE1 HIS A 64 1.310 -11.573 7.431 1.00 0.00 H
TER 1133 HIS A 64
ENDMDL
MODEL 3
ATOM 1 N MET A 1 12.132 -8.125 2.725 1.00 0.00 N
ATOM 2 CA MET A 1 11.117 -7.267 2.133 1.00 0.00 C
ATOM 3 C MET A 1 11.223 -7.328 0.620 1.00 0.00 C
...
となっており、Model 1〜25まで存在することがわかります。一方でX線結晶構造解析法で決められたデータには通常複数のModelはなく、1つのStructureオブジェクトに1つのModel になっています。
BioPythonを使って所望のデータを取り出す
Structure-Model-Chain-Residue-Atom(SMCRA)Frameworkのことを理解した上で、いくつかの例を示しながら、PDB構造ファイルから所望のデータを取り出すやり方を学んでいきます。
まずは構造ファイルを変数strucに代入してみるところから始めます。ここではPDB ID: 1EN2について、あえてPDBフォーマット形式でダウンロードしてみます。そして、mmCIFフォーマットではなくpdbフォーマットをパースして変数strucにロードするには、以下に指定されているようにBio.PDB.PDBParserを使用します。参考:https://biopython.org/docs/latest/api/Bio.PDB.PDBParser.html
from Bio.PDB.PDBList import PDBList
from Bio.PDB.PDBParser import PDBParser
pdbl = PDBList()
parser = PDBParser(QUIET=True)
pdbl.retrieve_pdb_file("1EN2", pdir="./", file_format="pdb", overwrite=True)
struc = parser.get_structure("1EN2", "pdb1en2.ent")
これでstrucにpdb1en2.entについての構造情報がロードされました。ちなみにファイル名pdb1en2.entは1en2.pdbという名前でPDBからダウンロードすることも可能で、両者は等価です。このpdb1en2.ent(または1en2.pdb)をVSCodeなど任意のテキストエディタで開き、ATOMレコード部分を確認してみると、450行目くらいから
CRYST1 31.820 39.600 63.640 90.00 90.00 90.00 P 21 21 21 4
ORIGX1 1.000000 0.000000 0.000000 0.00000
ORIGX2 0.000000 1.000000 0.000000 0.00000
ORIGX3 0.000000 0.000000 1.000000 0.00000
SCALE1 0.031427 0.000000 0.000000 0.00000
SCALE2 0.000000 0.025253 0.000000 0.00000
SCALE3 0.000000 0.000000 0.015713 0.00000
HETATM 1 N PCA A 1 0.525 2.690 13.317 1.00 20.26 N
HETATM 2 CA PCA A 1 -0.993 2.924 13.160 1.00 21.23 C
HETATM 3 CB PCA A 1 -1.783 1.879 14.043 1.00 24.73 C
HETATM 4 CG PCA A 1 -0.609 1.208 14.621 1.00 22.31 C
HETATM 5 CD PCA A 1 0.869 1.560 14.229 1.00 23.27 C
HETATM 6 OE PCA A 1 2.053 1.323 14.293 1.00 26.24 O
HETATM 7 C PCA A 1 -1.375 4.270 13.719 1.00 19.21 C
HETATM 8 O PCA A 1 -0.389 5.021 14.230 1.00 18.05 O
ATOM 9 N ARG A 2 -2.607 4.673 13.504 1.00 20.57 N
ATOM 10 CA ARG A 2 -3.091 6.010 13.918 1.00 20.82 C
ATOM 11 C ARG A 2 -3.361 6.101 15.403 1.00 19.55 C
ATOM 12 O ARG A 2 -3.742 5.121 16.050 1.00 19.99 O
ATOM 13 CB ARG A 2 -4.348 6.395 13.124 1.00 25.30 C
ATOM 14 CG ARG A 2 -4.110 6.337 11.619 1.00 30.98 C
ATOM 15 CD ARG A 2 -4.277 7.652 10.942 1.00 31.79 C
ATOM 16 NE ARG A 2 -3.441 8.730 11.457 1.00 31.91 N
ATOM 17 CZ ARG A 2 -3.550 9.979 10.980 1.00 31.95 C
ATOM 18 NH1 ARG A 2 -4.444 10.212 10.022 1.00 33.34 N
ATOM 19 NH2 ARG A 2 -2.804 10.949 11.444 1.00 29.59 N
ATOM 20 N CYS A 3 -3.248 7.317 15.948 1.00 15.71 N
ATOM 21 CA CYS A 3 -3.419 7.555 17.361 1.00 14.13 C
ATOM 22 C CYS A 3 -3.637 9.026 17.672 1.00 16.22 C
ATOM 23 O CYS A 3 -3.406 9.901 16.828 1.00 15.46 O
ATOM 24 CB CYS A 3 -2.127 7.069 18.110 1.00 13.74 C
ATOM 25 SG CYS A 3 -0.659 7.917 17.525 1.00 11.51 S
ATOM 26 N GLY A 4 -4.122 9.328 18.863 1.00 15.45 N
ATOM 27 CA GLY A 4 -4.129 10.656 19.402 1.00 17.05 C
ATOM 28 C GLY A 4 -5.029 11.674 18.735 1.00 16.11 C
ATOM 29 O GLY A 4 -6.039 11.345 18.125 1.00 17.88 O
ATOM 30 N SER A 5 -4.651 12.958 18.899 1.00 17.09 N
ATOM 31 CA SER A 5 -5.559 14.034 18.479 1.00 19.08 C
となっていることがわかります。
さて、ここは練習としてModel 1のChain Aの残基番号3のアミノ酸残基の名前を変数aに、残基番号3のアミノ酸残基のCα原子のxyz座標を変数bにリスト形式で返すコマンド(ついでに型の確認も)について紹介します。つまり、aには"CYS"が、bにはATOM 21の行に存在する座標[-3.419, 7.555, 17.361]がそれぞれ代入されることを期待してプログラムをかければいいわけですね。
これをBioPythonを使って書くと、以下のようにそれぞれ簡単にかけます。
from Bio.PDB.PDBList import PDBList
from Bio.PDB.PDBParser import PDBParser
# Bio.PDBのサブモジュールBio.PDB.AtomのAtomクラスをインポート
from Bio.PDB.Atom import Atom
# Bio.PDBのサブモジュールBio.PDB.ResidueのResidueクラスをインポート
from Bio.PDB.Residue import Residue
pdbl = PDBList()
parser = PDBParser(QUIET=True)
pdbl.retrieve_pdb_file("1EN2", pdir="./", file_format="pdb", overwrite=True)
struc = parser.get_structure("1EN2", "pdb1en2.ent")
# 最初のModel 1(Pythonは0はじまりなので、[0])、Chain A(["A"])、残基番号3のアミノ酸([3], アミノ酸の残基番号は0基準でない)の残基の名前を返すメソッドget_resname()を使う
print(
type(struc[0]["A"][3])
) # <class 'Bio.PDB.Residue.Residue'>と表示される。Residueオブジェクトであることを表す。
a = Residue.get_resname(struc[0]["A"][3])
print(a) # CYSと表示される
print(type(a)) # <class 'str'>と表示される。aの型はString型である。
print(
type(struc[0]["A"][3]["CA"])
) # <class 'Bio.PDB.Atom.Atom'>と表示される。Atomオブジェクトであることを表す。
# Atomクラスのメソッドget_coordを用いて、上記残基番号3のCYSのCα原子のxyz座標をリスト形式で返す
b = Atom.get_coord(struc[0]["A"][3]["CA"])
print(b) # [-3.419 7.555 17.361]と表示される
print(type(b)) # <class 'numpy.ndarray'> と表示される。bの型はNumPy配列型である。
https://biopython.org/docs/latest/api/Bio.PDB.Atom.html
まず前半については、struc[0]["A"]["3"]を入力するとBio.PDB.Residueの定義するResidueオブジェクトが発生しており、それに対してBio.PDB.Residueのメソッド(= 各クラスについて専用に定義された関数のこと)get_resname()を適用することで、その残基名を取り出しています。同様にして後半について、struc[0]["A"]["3"]["CA"]によって得られるAtomオブジェクトについて、$x, y, z$座標を取り出すメソッドget_coord()を適用しています。
ヘッダー情報の取得
PDBフォーマットの場合
PDBParserを使って変数に格納されたPDBファイル(not mmCIFファイル)には、タンパク質の構造情報だけでなく、様々な付随情報も含まれています。これらの付随情報はヘッダー情報と呼ばれており、PDBフォーマットではファイルの先頭からATOMレコードが始まるまでのHEADER行、REMARK行に含まれています。
Bio.PDBのParserで格納されたファイルのヘッダー情報はPython文法の辞書(dict)形式で格納されているため、keyを指定すると対応したvalueが取り出せるという形式になっています。例として先程の1EN2についてヘッダー情報を調べてみましょう。まずすべてのヘッダー情報に含まれる辞書のkey一覧を取得するために、変数struc.header.keys()を実行します。
from Bio.PDB.PDBList import PDBList
from Bio.PDB.PDBParser import PDBParser
pdbl = PDBList()
parser = PDBParser(QUIET=True)
pdbfile = "pdb1en2.ent"
struc = parser.get_structure("1EN2", pdbfile)
print(struc.header.keys())
# 結果
# >>> dict_keys(['name', 'head', 'idcode', 'deposition_date', 'release_date', 'structure_method', 'resolution', 'structure_reference', 'journal_reference', 'author', 'compound', 'source', 'has_missing_residues', 'missing_residues', 'keywords', 'journal'])
これで存在するkeysが調べられましたので、それぞれのkeyを使ってその中の値を取り出す操作を実行してみましょう。
# PDBへの登録名。
>>> print(struc.header["name"])
UDA TETRASACCHARIDE COMPLEX. CRYSTAL STRUCTURE OF URTICA DIOICA AGGLUTININ, A SUPERANTIGEN PRESENTED BY MHC MOLECULES OF CLASS I AND CLASS II
# 1行での説明。PDBフォーマットの場合は1行目のHEADERの記述に相当。またはClassificationとも。
>>> print(struc.header["head"])
SUGAR BINDING PROTEIN
# 構造のPDB ID
>>> print(struc.header["idcode"])
1EN2
# 構造のProtein Data Bankへの登録日
>>> print(struc.header["deposition_date"])
2000-03-20
# 構造の一般公開日
>>> print(struc.header["release_date"])
2000-06-21
# 構造決定の手法。多くの場合、X線結晶構造解析、NMR、クライオ電子顕微鏡のいずれか。
>>> print(struc.header["structure_method"])
X-RAY DIFFRACTION
# 結晶構造解析の分解能(単位はÅ)。結晶構造(X-RAY DIFFRACTION)の場合のみ存在する。
>>> print(struc.header["resolution"])
1.4
(以下略)
このように、ヘッダーに登録されている構造以外の付随情報も取り出すことができます。
しかしながら、多くのPDBフォーマットについてはヘッダー情報が不完全だったり、エラーを含んでいる場合があります。それらの多くのエラーは、現行のmmCIFフォーマットに情報を移植するときに修正されています。したがって、もし構造ファイルからヘッダー情報を取り出したい場合は、PDBフォーマットのヘッダー情報を取り出すよりも、Bio.PDB.MMCIF2Ditを使って情報を取り出すほうが正確なため、こちらを使うことを勧めます。
mmCIFフォーマットの場合
Bio.PDB.MMCIF2Dictモジュールを使ってmmCIFファイルのデータをdictに入れ込みます。
参考: https://biopython.org/docs/latest/api/Bio.PDB.MMCIF2Dict.html
import gzip
import urllib.request
from mimetypes import guess_type
from Bio.PDB.MMCIF2Dict import MMCIF2Dict
ciffile, httpobject = urllib.request.urlretrieve(
"https://files.rcsb.org/download/1en2.cif.gz", "1en2.cif.gz"
)
# gzip圧縮/非圧縮、両対応の_open関数
def _open(file):
encoding = guess_type(file)[1]
if encoding == "gzip":
return gzip.open(file, mode="rt")
else:
return open(file)
ciffile = "1en2.cif.gz" # gzip圧縮でないmmCIFファイルでも可
with _open(ciffile) as handle:
# ここでmmCIFの情報をdictのkey, valueに変換する
mmcifdict = MMCIF2Dict(handle)
print(mmcifdict)
こちらはPDBフォーマットの場合と違って大量にprint結果が表示されます。あまりに大量にあるので適切なkeyを探すのは大変ですが、 https://mmcif.wwpdb.org/dictionaries/mmcif_pdbx_v50.dic/Items/ のウェブサイトで目的のkeyを調べると便利です。
以下では適切なkeyを入力することで、上記のPDBフォーマットの結果と等価な結果を返すことができることを示してみます。多くの場合はリスト型になっているので、データの取り扱いには気をつけましょう。
# PDBへの登録名。
>>> print(mmcifdict["_struct.title"])
['UDA TETRASACCHARIDE COMPLEX. CRYSTAL STRUCTURE OF URTICA DIOICA AGGLUTININ, A SUPERANTIGEN PRESENTED BY MHC MOLECULES OF CLASS I AND CLASS II']
# 1行での説明。PDBフォーマットの場合は1行目のHEADERの記述に相当。またはClassificationとも。
>>> print(mmcifdict["_struct_keywords.pdbx_keywords"])
['SUGAR BINDING PROTEIN']
# 構造のPDB ID
>>> print(mmcifdict["_struct.entry_id"])
['1EN2']
# 構造のProtein Data Bankへの登録日
>>> print(mmcifdict["_pdbx_database_status.recvd_initial_deposition_date"])
['2000-03-20']
# 構造の一般公開日(→これはデータ上での変更を加えた日(revision date)に変更されている)
>>> print(mmcifdict["_pdbx_audit_revision_history.revision_date"])
['2000-06-21', '2008-04-27', '2011-07-13', '2014-02-05', '2019-12-25', '2020-07-29']
# 構造の一般公開日(revision ver.1.0の公開日)
>>> print(mmcifdict["_pdbx_audit_revision_history.revision_date"][0])
2000-06-21
# 構造決定の手法。多くの場合、X線結晶構造解析、NMR、クライオ電子顕微鏡のいずれか。
>>> print(mmcifdict["_exptl.method"])
['X-RAY DIFFRACTION']
# 結晶構造解析の分解能(単位はÅ)。結晶構造(X-RAY DIFFRACTION)の場合のみ存在する。
>>> print(mmcifdict["_reflns.d_resolution_high"])
['1.4']
データ構造の操作
上述のようにPDBの生体分子構造情報はEntityを最上とするSMCRAデータフレームワークとなっていることから、Structure, Model, Chain, Residue, Atomの各要素(エレメント)は以下のようにループさせて取り出すことができます。またModelから見てChain, residue, atomのエレメントは下位に存在するためにModelのサブエレメントと呼びます。同様にChainに対するresidueやatomエレメント, residueに対するatomエレメントはサブエレメントとなります。
for model in structure.get_list():
for chain in model.get_list():
for residue in chain.get_list():
for atom in residue.get_list():
# Do something
PDB ID: 1EN2について、residueエレメントをすべて表示する以下のPythonスクリプトを実行してみます。
import gzip
from mimetypes import guess_type
from Bio.PDB.MMCIFParser import FastMMCIFParser
# gzip圧縮/非圧縮、両対応の_open関数
def _open(file):
encoding = guess_type(file)[1]
if encoding == "gzip":
return gzip.open(file, mode="rt")
else:
return open(file)
ciffile = "1en2.cif.gz"
pdb_parser = FastMMCIFParser(QUIET=True)
with _open(ciffile) as handle:
struc = pdb_parser.get_structure("1EN2", handle)
for model in struc.get_list():
for chain in model.get_list():
for residue in chain.get_list():
print(residue)
結果はこうなります
<Residue PCA het=H_PCA resseq=1 icode= >
<Residue ARG het= resseq=2 icode= >
<Residue CYS het= resseq=3 icode= >
<Residue GLY het= resseq=4 icode= >
<Residue SER het= resseq=5 icode= >
<Residue GLN het= resseq=6 icode= >
<Residue GLY het= resseq=7 icode= >
<Residue GLY het= resseq=8 icode= >
<Residue GLY het= resseq=9 icode= >
<DisorderedResidue GLY het= resseq=10 icode= >
<Residue THR het= resseq=11 icode= >
<Residue CYS het= resseq=12 icode= >
<Residue PRO het= resseq=13 icode= >
<DisorderedResidue ALA het= resseq=14 icode= >
<Residue LEU het= resseq=15 icode= >
<DisorderedResidue TRP het= resseq=16 icode= >
<Residue CYS het= resseq=17 icode= >
<Residue CYS het= resseq=18 icode= >
<Residue SER het= resseq=19 icode= >
...
(中略)
...
<Residue GLY het= resseq=79 icode= >
<DisorderedResidue SER het= resseq=80 icode= >
<DisorderedResidue LYS het= resseq=81 icode= >
<Residue CYS het= resseq=82 icode= >
<Residue GLN het= resseq=83 icode= >
<Residue TYR het= resseq=84 icode= >
<Residue ARG het= resseq=85 icode= >
<Residue CYS het= resseq=86 icode= >
<Residue HOH het=H_HOH resseq=94 icode= >
<Residue HOH het=H_HOH resseq=95 icode= >
<Residue HOH het=H_HOH resseq=96 icode= >
<Residue HOH het=H_HOH resseq=97 icode= >
<Residue HOH het=H_HOH resseq=98 icode= >
<Residue HOH het=H_HOH resseq=99 icode= >
<Residue HOH het=H_HOH resseq=100 icode= >
<Residue HOH het=H_HOH resseq=101 icode= >
<Residue HOH het=H_HOH resseq=102 icode= >
(後略)
この結果は、それぞれの残基がResidueオブジェクトとして認識されていることを表しています。このうち、標準の20種類のアミノ酸以外の残基の場合(残基名PCAやHOHなど)は ヘテロ残基 (Hetero residue) として認識されます。このときhet=の後にH_という接頭辞がついて区別されます。
Structure、Model、Chain、ResidueはすべてEntityベースクラスのサブクラスです。Atomクラスは、Entityインターフェースを(部分的に)実装しているだけです(Atomは子を持たないため)。
各Entityサブクラスでは、その子の一意のIDをkeyとして子を抽出することができます(例: 原子名の文字列をkeyとしてResidueオブジェクトからAtomオブジェクトを抽出することができ、Chainの識別子をkeyとしてModelオブジェクトからChainオブジェクトを抽出することができます)。
Disordered atomやresidueは、DisorderedEntityWrapperベースクラスのサブクラスであるDisorderedAtomクラスやDisorderedResidueクラスで表現されます。DisorderedAtomとDisorderedResidueクラスは、DisorderedEntityWrapperベースクラスのサブクラスです。
一般的に、子のEntityオブジェクト(Atom、Residue、Chain、Model)は、idをキーにして親(Residue、Chain、Model、Structureそれぞれ)から抽出することができます。
>>> child_entity = parent_entity[child_id]
>>> # 具体例: StructureのModel 0のデータを`child_entity`という変数に入れる。
>>> child_entity = struc[0]
>>> # このchild_entityはstructureオブジェクトの子なので、Modelオブジェクト扱いになっている
>>> type(child_entity)
<class 'Bio.PDB.Model.Model'>
>>> # 具体例: StructureのModel 0のChain Aのデータを`child_entity`という変数に入れる。
>>> child_entity = struc[0]["A"]
>>> # このchild_entityはModelオブジェクトの子なので、Chainオブジェクト扱いになっている
>>> type(child_entity)
<class 'Bio.PDB.Chain.Chain'>
また、メソッド.get_list()を使うことで親Entityオブジェクトのすべての子Entityのリストを取得することもできます。このリストは特定の方法でソートされていることに注意してください(例:Modelオブジェクト中のChainオブジェクトの場合、Chain識別子に従ってソートされています)。
>>> child_list = parent_entity.get_list()
>>> # 具体例: StructureのModel 0のデータの全ての子オブジェクト(chainオブジェクト)のリスト。
>>> child_list = struc[0].get_list()
>>> print(child_list)
[<Chain id=A>, <Chain id=B>]
>>> # 具体例: StructureのModel 0のChain Aのデータの全ての子オブジェクト(Residueオブジェクト)のリスト。
>>> child_list = struc[0]["A"].get_list()
>>> print(child_list)
[<Residue PCA het=H_PCA resseq=1 icode= >, <Residue ARG het= resseq=2 icode= >, ... ]
反対に、子オブジェクトから親オブジェクトの情報を得る事もできます。
>>> parent_entity = child_entity.get_parent()
>>> # 具体例: StructureのModel 0のChain Aの親オブジェクトを返す(→Model 0になるはず)。
>>> parent_entity = struc[0]["A"].get_parent()
>>> print(parent_entity)
<Model id=0>
SMCRA階層構造のすべてのレベルにおいて、メソッド.get_full_id()を使うことでfull idを取得することができます。full idとはトップ階層のオブジェクト(Structure)から始まって現在のオブジェクトに下るまでのすべてのidを含んだtuple形式になっています。例えば、Residueオブジェクトについてfull idの取得を試みるとこのようになります:
>>> ResidueArg2 = struc[0]["A"][2]
>>> print(ResidueArg2.get_full_id())
('1EN2', 0, 'A', (' ', 2, ' '))
左から順にStructure ID, Model ID, Chain ID, Residue ID (Residueレベルのみ三つ組記法となっている。これについては後述。)となったtuple形式になっています。
次にStructure, Model, Chain, Residue, Atomオブジェクトそれぞれについて詳細に解説していきます。
Structure
Structureオブジェクトは階層の最上位に位置します。そのidは、ユーザーが指定した文字列です。Structureには、いくつかのModelの子オブジェクト(child object)が含まれています。ほとんどの結晶構造(すべてではありません)は単一のモデルを含みますが、NMR構造は通常、複数のモデルで構成されます。また、分子の大部分の結晶構造が乱れている場合にも、複数のモデルが存在することがあります。
Model
Modelオブジェクトのidは整数で,解析されたファイルの中でのモデルの位置から導き出されます(0から始まる自動的な番号が付けられます)。結晶構造は通常1つのモデル(id 0)しか持ちませんが、NMRファイルは通常複数のモデルを持ちます。多くのPDBパーサはModelが1つであることを前提としていますが、Bio.PDBのStructureクラスは、複数のモデルを持つPDBファイルを簡単に扱えるように設計されています。例えば、Structureオブジェクトから最初のModelを取得するには、
>>> first_model = struc[0]
Modelオブジェクトは子オブジェクトとしてChainのリストを格納しています。
Chain
Chainオブジェクトのidは、PDB/mmCIFファイルのchainの識別子に由来するもので、1文字(通常はアルファベット1文字)です。モデル・オブジェクト内の各Chainは、ユニークなIDを持っています。例として、モデル・オブジェクトから識別子AのChainオブジェクトを取得するには、以下のようにします。
>>> chain_A = model["A"]
# first_modelのchain Aを取得したい場合は以下のようにも書ける
>>> chain_A = struc[0]["A"]
Chainオブジェクトは子オブジェクトとしてResidueのリストを格納しています。
Residue
Model, Chainと違って、Residueオブジェクトはちょっと複雑です。Residueのidは3つ組の要素のtupleになっています。
-
ヘテロフィールド (hetfield)
- 水分子の場合は
Wが付きます。(ただH_HOHで水分子を表している場合も多い) -
H_から続く残基名はヘテロ残基です。例えばグルコース分子を表すのにH_GLCとなっています。 - このヘテロフィールドは通常の20種類のアミノ酸や核酸の場合はブランク(
" ")となっています。
- 水分子の場合は
-
配列識別子 (sequence identifier) (resseq), chainの中での残基の位置を記述する整数値。(e.g.,
100)0始まりではないことに注意。すなわち、resseq=1であれば残基番号は1番である。これはPDBデータにおける残基番号は1番から始まらなかったり、途中欠損があったりするため、内部処理的には辞書のkey扱いになっているためである。 -
挿入コード(insertion code) (icode); String型。(e.g.
A). 挿入コードは、とある望ましい残基番号スキームを維持するために使用されることがあります。例としてPDB:1EN2の中にあるSer80挿入変異体はGly80とAsn81残基の間に挿入されています。このとき、以下のような配列識別子と挿入コードを持ちます。
ATOM 606 O SER A 78 11.178 -10.437 27.814 1.00 35.05 O
ATOM 607 CB SER A 78 14.183 -10.362 26.725 1.00 35.08 C
ATOM 608 OG SER A 78 15.357 -9.643 26.373 1.00 38.49 O
ATOM 609 N AGLY A 79 11.026 -10.753 25.586 0.56 32.73 N
ATOM 610 N BGLY A 79 11.268 -11.086 25.671 0.44 33.24 N
ATOM 611 CA AGLY A 79 9.821 -11.505 25.585 0.56 32.16 C
ATOM 612 CA BGLY A 79 10.123 -11.952 25.726 0.44 32.77 C
ATOM 613 C AGLY A 79 9.052 -11.575 26.887 0.56 32.04 C
ATOM 614 C BGLY A 79 9.866 -12.542 27.101 0.44 32.18 C
ATOM 615 O AGLY A 79 8.312 -10.654 27.235 0.56 32.01 O
ATOM 616 O BGLY A 79 10.734 -13.176 27.699 0.44 32.81 O
ATOM 617 N AGLY A 80 8.644 -12.355 27.599 0.44 31.90 N
ATOM 618 CA AGLY A 80 8.263 -12.856 28.907 0.44 30.90 C
ATOM 619 C AGLY A 80 8.635 -11.879 30.017 0.44 29.74 C
ATOM 620 O AGLY A 80 7.877 -11.725 30.978 0.44 31.09 O
ATOM 621 N BSER A 80 9.205 -12.682 27.605 0.56 31.46 N
ATOM 622 CA BSER A 80 8.473 -12.954 28.816 0.56 31.14 C
ATOM 623 C BSER A 80 8.880 -12.084 29.993 0.56 30.28 C
ATOM 624 O BSER A 80 8.341 -12.253 31.098 0.56 30.92 O
ATOM 625 CB BSER A 80 8.629 -14.441 29.205 0.56 32.24 C
ATOM 626 OG BSER A 80 9.999 -14.810 29.194 0.56 33.96 O
ATOM 627 N AASN A 81 9.796 -11.250 29.900 0.44 28.30 N
ATOM 628 CA AASN A 81 10.254 -10.303 30.910 0.44 26.39 C
ATOM 629 C AASN A 81 9.566 -8.946 30.756 0.44 24.18 C
ATOM 630 O AASN A 81 9.603 -8.128 31.673 0.44 23.07 O
ATOM 631 CB AASN A 81 11.773 -10.095 30.784 0.44 27.87 C
ATOM 632 CG AASN A 81 12.573 -11.218 31.412 0.44 29.21 C
ATOM 633 OD1AASN A 81 12.064 -11.941 32.265 0.44 28.74 O
ATOM 634 ND2AASN A 81 13.822 -11.353 30.992 0.44 30.49 N
ATOM 635 N BLYS A 81 9.846 -11.202 29.794 0.56 29.23 N
ATOM 636 CA BLYS A 81 10.291 -10.308 30.869 0.56 28.16 C
ATOM 637 C BLYS A 81 9.575 -8.959 30.759 0.56 26.65 C
ATOM 638 O BLYS A 81 9.248 -8.335 31.767 0.56 24.90 O
ATOM 639 CB BLYS A 81 11.795 -10.109 30.820 0.56 29.85 C
ATOM 640 CG BLYS A 81 12.626 -11.336 31.168 0.56 32.07 C
ATOM 641 CD BLYS A 81 14.097 -11.094 30.837 0.56 33.72 C
ATOM 642 CE BLYS A 81 14.890 -12.388 30.924 0.56 35.00 C
ATOM 643 NZ BLYS A 81 16.264 -12.237 30.375 0.56 35.39 N
ATOM 644 N CYS A 82 8.950 -8.719 29.607 1.00 22.45 N
ATOM 645 CA CYS A 82 8.458 -7.382 29.254 1.00 21.45 C
ATOM 646 C CYS A 82 6.972 -7.304 29.058 1.00 22.09 C
ATOM 647 O CYS A 82 6.378 -8.160 28.372 1.00 22.87 O
ATOM 648 CB CYS A 82 9.172 -6.970 27.942 1.00 18.44 C
このようにすることで、残基のナンバリングスキームにおいて、野生型(本来のすがた)の構造データと点変異が入った構造データを1ファイル中で共存させることができます。
以上のルールから、上記のグルコース残基のidは(’H GLC’, 100, ’A’)となります。hetero-flagとinsertion codeがともにブランク(" ")であるときならば、sequence識別子は以下のように省略して書くことができます。
# full id表記
>>> residue=chain[(" ", 100, " ")]
# 簡単なid表記(hetero-flag, icodeがブランクのときのみ動作)
>>> residue=chain[100]
Atom
Atomオブジェクトは、原子に関連するデータを格納するもので、子オブジェクトはありません。原子のIDは、その原子名です(例:Ser残基の側鎖の酸素原子を表す"OG")。原子のIDは、Residue内で一意である必要があります。DisorderedAtomについては例外があるため、ここではいったんないものとして考えてください。
原子のIDは単に原子の名前(例:CA)です。実際には、PDBファイルの原子名からスペースを全て取り除いて原子名を作成します。しかし、PDBファイルではスペースを原子名の一部として使用することができます。例えばカルシウム原子を'CA..'(ここで.はスペースの代わり)と呼ぶのは、主鎖のCα原子('.CA.'と呼ばれる)と区別するためであることが多いです。スペースを削除すると問題が生じる場合(例えば、同じ残基に'CA'と呼ばれる原子が2つある場合)には、スペースを残しています(とはいえ、発生する頻度はとても低いですが)。
保存される原子データは、原子名、原子座標(標準偏差がある場合はそれも含む)、B因子(異方性B因子と標準偏差がある場合はそれも含む)、altloc指定子、空白を含む完全な原子名です。PDBファイルで時折指定されることがあるもののあまり使われない項目(原子の元素番号や原子の電荷など)は保存されません。
原子座標を操作するには、Atomオブジェクトのtransform()メソッドを使用します。原子座標を直接指定するには、set_coord()メソッドを使用します。Atomオブジェクトには、以下の追加メソッドがあります。
DisorderedAtomとDisorderedResidue
遭遇する確率は低いですが、DisorderedAtomとDisorderedResidueについても説明します。DisorderedAtomの定義は同一原子だが、複数の異なる座標を持つデータで、DisorderedAtomの定義は同じ残基位置だが、複数の異なるアミノ酸残基を持つデータ(例として点変異など) のことです。PDBフォーマット上でこれを確認してみます。
ATOM 597 N CYS A 77 13.208 -5.961 28.876 1.00 22.81 N
ATOM 598 CA CYS A 77 12.814 -5.846 27.491 1.00 23.23 C
ATOM 599 C CYS A 77 12.573 -7.119 26.727 1.00 27.50 C
ATOM 600 O CYS A 77 11.961 -7.054 25.632 1.00 27.57 O
ATOM 601 CB CYS A 77 13.893 -5.011 26.744 1.00 21.70 C
ATOM 602 SG CYS A 77 13.970 -3.285 27.311 1.00 19.69 S
ATOM 603 N SER A 78 13.001 -8.275 27.211 1.00 29.47 N
ATOM 604 CA SER A 78 12.928 -9.511 26.448 1.00 32.50 C
ATOM 605 C SER A 78 11.665 -10.314 26.666 1.00 33.17 C
ATOM 606 O SER A 78 11.178 -10.437 27.814 1.00 35.05 O
ATOM 607 CB SER A 78 14.183 -10.362 26.725 1.00 35.08 C
ATOM 608 OG SER A 78 15.357 -9.643 26.373 1.00 38.49 O
ATOM 609 N AGLY A 79 11.026 -10.753 25.586 0.56 32.73 N
ATOM 610 N BGLY A 79 11.268 -11.086 25.671 0.44 33.24 N
ATOM 611 CA AGLY A 79 9.821 -11.505 25.585 0.56 32.16 C
ATOM 612 CA BGLY A 79 10.123 -11.952 25.726 0.44 32.77 C
ATOM 613 C AGLY A 79 9.052 -11.575 26.887 0.56 32.04 C
ATOM 614 C BGLY A 79 9.866 -12.542 27.101 0.44 32.18 C
ATOM 615 O AGLY A 79 8.312 -10.654 27.235 0.56 32.01 O
ATOM 616 O BGLY A 79 10.734 -13.176 27.699 0.44 32.81 O
ATOM 617 N AGLY A 80 8.644 -12.355 27.599 0.44 31.90 N
ATOM 618 CA AGLY A 80 8.263 -12.856 28.907 0.44 30.90 C
ATOM 619 C AGLY A 80 8.635 -11.879 30.017 0.44 29.74 C
ATOM 620 O AGLY A 80 7.877 -11.725 30.978 0.44 31.09 O
ATOM 621 N BSER A 80 9.205 -12.682 27.605 0.56 31.46 N
ATOM 622 CA BSER A 80 8.473 -12.954 28.816 0.56 31.14 C
ATOM 623 C BSER A 80 8.880 -12.084 29.993 0.56 30.28 C
ATOM 624 O BSER A 80 8.341 -12.253 31.098 0.56 30.92 O
ATOM 625 CB BSER A 80 8.629 -14.441 29.205 0.56 32.24 C
ATOM 626 OG BSER A 80 9.999 -14.810 29.194 0.56 33.96 O
ATOM 627 N AASN A 81 9.796 -11.250 29.900 0.44 28.30 N
ATOM 628 CA AASN A 81 10.254 -10.303 30.910 0.44 26.39 C
ATOM 629 C AASN A 81 9.566 -8.946 30.756 0.44 24.18 C
ATOM 630 O AASN A 81 9.603 -8.128 31.673 0.44 23.07 O
ATOM 631 CB AASN A 81 11.773 -10.095 30.784 0.44 27.87 C
ATOM 632 CG AASN A 81 12.573 -11.218 31.412 0.44 29.21 C
ATOM 633 OD1AASN A 81 12.064 -11.941 32.265 0.44 28.74 O
ATOM 634 ND2AASN A 81 13.822 -11.353 30.992 0.44 30.49 N
ATOM 635 N BLYS A 81 9.846 -11.202 29.794 0.56 29.23 N
ATOM 636 CA BLYS A 81 10.291 -10.308 30.869 0.56 28.16 C
ATOM 637 C BLYS A 81 9.575 -8.959 30.759 0.56 26.65 C
ATOM 638 O BLYS A 81 9.248 -8.335 31.767 0.56 24.90 O
ATOM 639 CB BLYS A 81 11.795 -10.109 30.820 0.56 29.85 C
ATOM 640 CG BLYS A 81 12.626 -11.336 31.168 0.56 32.07 C
ATOM 641 CD BLYS A 81 14.097 -11.094 30.837 0.56 33.72 C
ATOM 642 CE BLYS A 81 14.890 -12.388 30.924 0.56 35.00 C
ATOM 643 NZ BLYS A 81 16.264 -12.237 30.375 0.56 35.39 N
ATOM 644 N CYS A 82 8.950 -8.719 29.607 1.00 22.45 N
ATOM 645 CA CYS A 82 8.458 -7.382 29.254 1.00 21.45 C
ATOM 646 C CYS A 82 6.972 -7.304 29.058 1.00 22.09 C
ATOM 647 O CYS A 82 6.378 -8.160 28.372 1.00 22.87 O
ATOM 648 CB CYS A 82 9.172 -6.970 27.942 1.00 18.44 C
ATOM 649 SG CYS A 82 8.735 -5.286 27.433 1.00 17.39 S
このPDB:1EN2のデータの例では、Chain A(22列目の文字)の79番目(23-26列目の数字)のグリシン残基(18-20列目の文字)はすべての原子(N,CA,C,O)について17列目にAlternative location indicatorであることを示すAまたはBの文字が付与されており、本来1つであるはずの構成原子の位置に複数の座標データが含まれていることがわかります。同様に、80番目の残基を見ると、今度はグリシン残基とセリン残基が同時にデータ上存在していることがわかります。なんとも奇妙で、特に2つの残基が同時に存在するDisorderedResidueはほとんど現実にはありえませんが、データ上は存在しうると思ってください。一方である原子について残基名・残基番号が同じにも関わらず2つ以上の座標が存在しうるDisorderedAtomはそこそこ高確率で見かけます。
ある対象原子または残基がそれぞれDisorderedAtom, DisorderedResidueであるかどうかを0または1(0がFalse, 1がTrueに相当する)で返す関数はAtom.is_disordered(atom)とResidue.is_disordered(residue)を使います。
import gzip
from mimetypes import guess_type
import Bio.PDB
import Bio.PDB.Atom
import Bio.PDB.Residue
from Bio.PDB.MMCIFParser import FastMMCIFParser
# gzip圧縮/非圧縮、両対応の_open関数
def _open(file):
encoding = guess_type(file)[1]
if encoding == "gzip":
return gzip.open(file, mode="rt")
else:
return open(file)
ciffile = "1en2.cif.gz"
pdb_parser = FastMMCIFParser()
with _open(ciffile) as handle:
struc = pdb_parser.get_structure("1EN2", handle)
# 1EN2のChain Aの78番目の残基のCA原子がDisorderedAtomであるかを0または1で返す関数
print(Bio.PDB.Atom.Atom.is_disordered(struc[0]["A"][78]["CA"])) # 0
# 1EN2のChain Aの79番目の残基のCA原子がDisorderedAtomであるかを0または1で返す関数
print(Bio.PDB.Atom.Atom.is_disordered(struc[0]["A"][79]["CA"])) # 1
print(Bio.PDB.Atom.Atom.is_disordered(struc[0]["A"][80]["CA"])) # 1
print(Bio.PDB.Atom.Atom.is_disordered(struc[0]["A"][81]["CA"])) # 1
print(Bio.PDB.Atom.Atom.is_disordered(struc[0]["A"][82]["CA"])) # 0
# 1EN2のChain Aの78番目の残基がDisorderedResidueであるかを0または1で返す関数
print(Bio.PDB.Residue.Residue.is_disordered(struc[0]["A"][78])) # 0
print(Bio.PDB.Residue.Residue.is_disordered(struc[0]["A"][79])) # 1
print(Bio.PDB.Residue.Residue.is_disordered(struc[0]["A"][80])) # 1
print(Bio.PDB.Residue.Residue.is_disordered(struc[0]["A"][81])) # 1
print(Bio.PDB.Residue.Residue.is_disordered(struc[0]["A"][82])) # 0
返り値はint型で0または1になっているので、if文の真偽判定の条件式に使うことができます。
# num = 80ならばdisordered, num = 78ならばNOT disorderedと表示されます。
num = 80
if(Bio.PDB.Residue.Residue.is_disordered(struc[0]["A"][num])):
print("Residue {0} is disordered.".format(num))
else:
print("Residue {0} is NOT disordered.".format(num))
また、DisorderedResidueについては、通常1つのデータのみが自動で取得され、残りのデータについては無視されます。
>>> ciffile = "1en2.cif"
>>>
>>> pdb_parser = Bio.PDB.FastMMCIFParser(QUIET=True)
>>> with open(ciffile) as handle:
... struc = pdb_parser.get_structure("1EN2", handle)
...
# PDB:1EN2について、残基番号80はGLY, SERの2つがあるが、この場合どうなるのか。
>>> res = struc[0]["A"][80]
>>> print(res.get_resname())
SER # 残基番号80はSERの方の情報だけが取得されている
>>> res = struc[0]["A"][81]
>>> print(res.get_resname())
LYS # 残基番号80はSERの方の情報だけが取得されている
1EN2のChain Aの残基番号80には、データ上はGLY80(A)とSER80(B)がありますが、Bの方だけが採用されています。続く残基番号81について同様に実行してみても、LYSの方しか表示されません。
一応、chainオブジェクトを取り出すときにメソッド.get_unpacked_list()をつけると、表示されない残りの残基の情報も取得することができるようになります。
>>> chain = struc[0]["A"].get_unpacked_list()
>>> chain[80]
<Residue SER het= resseq=78 icode= >
>>> struc[0]["A"].get_unpacked_list()[80] # 上に同じ
<Residue SER het= resseq=78 icode= >
>>> chain[81]
<Residue GLY het= resseq=79 icode= >
>>> chain[82]
<Residue GLY het= resseq=80 icode= >
>>> chain[83]
<Residue SER het= resseq=80 icode= >
>>> chain[84]
<Residue ASN het= resseq=81 icode= >
しかし、[ ]の中の引数とresseqが一致しなくなるので取り扱いが難しくなります。というか、多くの場合において見えないもう一方の方を利用したいという場面はとても少ないと思いますので、この現象は無視してもいいかもしれません。
解析に使える関数
Atomオブジェクトについて
例として、以下のコードを実行した状態にしておきます。
import gzip
from mimetypes import guess_type
import Bio.PDB
import Bio.PDB.Atom
import Bio.PDB.Residue
from Bio.PDB.MMCIFParser import FastMMCIFParser
# gzip圧縮/非圧縮、両対応の_open関数
def _open(file):
encoding = guess_type(file)[1]
if encoding == "gzip":
return gzip.open(file, mode="rt")
else:
return open(file)
ciffile = "1en2.cif.gz"
pdb_parser = FastMMCIFParser()
with _open(ciffile) as handle:
struc = pdb_parser.get_structure("1EN2", handle)
# Chain A 残基番号78のCα原子についてのAtomオブジェクト
at = struc[0]["A"][78]["CA"]
Atomオブジェクトについて使用できる各関数をいくつか実行してみます。
>>> at.get_name() # atom name (spaces stripped, e.g. ’CA’)
'CA'
>>> at.get_id() # id (equals atom name)
'CA'
>>> at.get_coord() # atomic coordinates
array([12.928, -9.511, 26.448], dtype=float32)
>>> at.get_vector() # atomic coordinates as Vector object
<Vector 12.93, -9.51, 26.45>
>>> at.get_bfactor() # isotropic B factor
32.5
>>> at.get_occupancy() # occupancy
1.0
>>> at.get_altloc() # alternative location specifier
' '
>>> at.get_sigatm() # std. dev. of atomic parameters
>>> at.get_siguij() # std. dev. of anisotropic B factor
>>> at.get_anisou() # anisotropic B factor
>>> at.get_fullname() # atom name (with spaces, e.g. ’.Cat.’)
'CA'
これらの各値は、pdbフォーマットのATOMレコードの読み方と合わせれば理解できるでしょう。
ATOM 603 N SER A 78 13.001 -8.275 27.211 1.00 29.47 N
ATOM 604 CA SER A 78 12.928 -9.511 26.448 1.00 32.50 C
ATOM 605 C SER A 78 11.665 -10.314 26.666 1.00 33.17 C
ATOM 606 O SER A 78 11.178 -10.437 27.814 1.00 35.05 O
ATOM 607 CB SER A 78 14.183 -10.362 26.725 1.00 35.08 C
ATOM 608 OG SER A 78 15.357 -9.643 26.373 1.00 38.49 O
他にも関数はたくさんありますので、公式ドキュメントを読んでください。
Residueオブジェクトについて
例として、以下のコードを実行した状態にしておきます。
import gzip
from mimetypes import guess_type
import Bio.PDB
import Bio.PDB.Atom
import Bio.PDB.Residue
from Bio.PDB.MMCIFParser import FastMMCIFParser
# gzip圧縮/非圧縮、両対応の_open関数
def _open(file):
encoding = guess_type(file)[1]
if encoding == "gzip":
return gzip.open(file, mode="rt")
else:
return open(file)
ciffile = "1en2.cif.gz"
pdb_parser = FastMMCIFParser()
with _open(ciffile) as handle:
struc = pdb_parser.get_structure("1EN2", handle)
# Chain A 残基番号78についてのResidueオブジェクト
res = struc[0]["A"][78]
Residueオブジェクトについて使用できる各関数をいくつか実行してみます。
>>> res.get_resname() # return the residue name (eg. ’GLY’)
'SER'
>>> res.is_disordered() # 1 if the residue has disordered atoms
0
>>> res.get_segid() # return the SEGID
' '
>>> res.has_id("CA") # test if a residue has a certain atom
True
>>> res.has_id("SG")
False
他にもたくさんありますので、公式ドキュメントを読んでください。
例1: 原子間距離を計算する
例として1EN2のChain AのTRP-21のNE1原子とChain BのNAG-1のO6原子間の距離を取得してみます。PyMOLで表示するとこんな感じです。
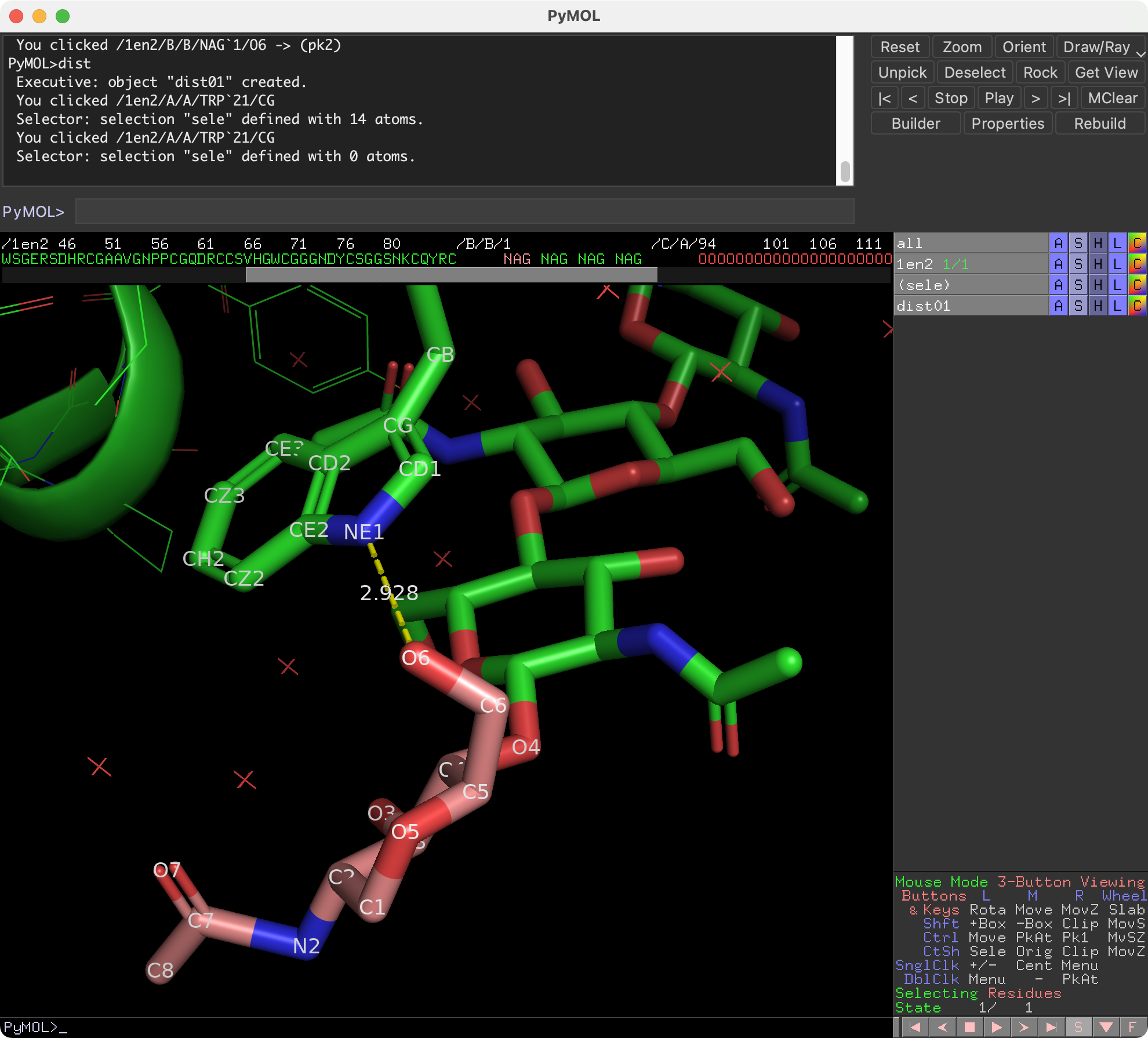
この2.928Åを取得してみたいと考えます。
Atomオブジェクトについて、マイナス演算子(-)が2点間の原子オブジェクトの距離を返すよう関数のオーバーロードがなされているため、以下のようにして原子間距離を簡単に計算することができます。
import gzip
import urllib.request
from mimetypes import guess_type
import Bio.PDB
import Bio.PDB.Atom
import Bio.PDB.Residue
from Bio.PDB.MMCIFParser import FastMMCIFParser
def _open(file):
encoding = guess_type(file)[1]
if encoding == "gzip":
return gzip.open(file, mode="rt")
else:
return open(file)
urllib.request.urlretrieve("https://files.rcsb.org/download/1en2.cif.gz", "1en2.cif.gz")
ciffile = "1en2.cif.gz"
# 1EN2構造をstrucにロード
pdb_parser = FastMMCIFParser()
with _open(ciffile) as handle:
struc = pdb_parser.get_structure("1EN2", handle)
# %%
# 1EN2構造中のChain AのTRP-21のNE1原子とChain BのNAG-1のO6原子間の距離を取得する
atomA = struc[0]["A"][21]["NE1"]
atomB = struc[0]["B"][("H_NAG", 1, " ")]["O6"]
distanceAB = atomA - atomB
# 距離の表示
print(distanceAB)
最後のprint文で変数distanceABに目的の距離情報が取得できていることがわかります。
>>> 2.9284787
例2: 3点の原子の作る角度を計算する
例として1EN2のChain AのTRP-21のCD1原子とNE1原子とChain BのNAG-1のO6原子が作る角度(°)を取得してみます。PyMOLで表示するとこんな感じです。
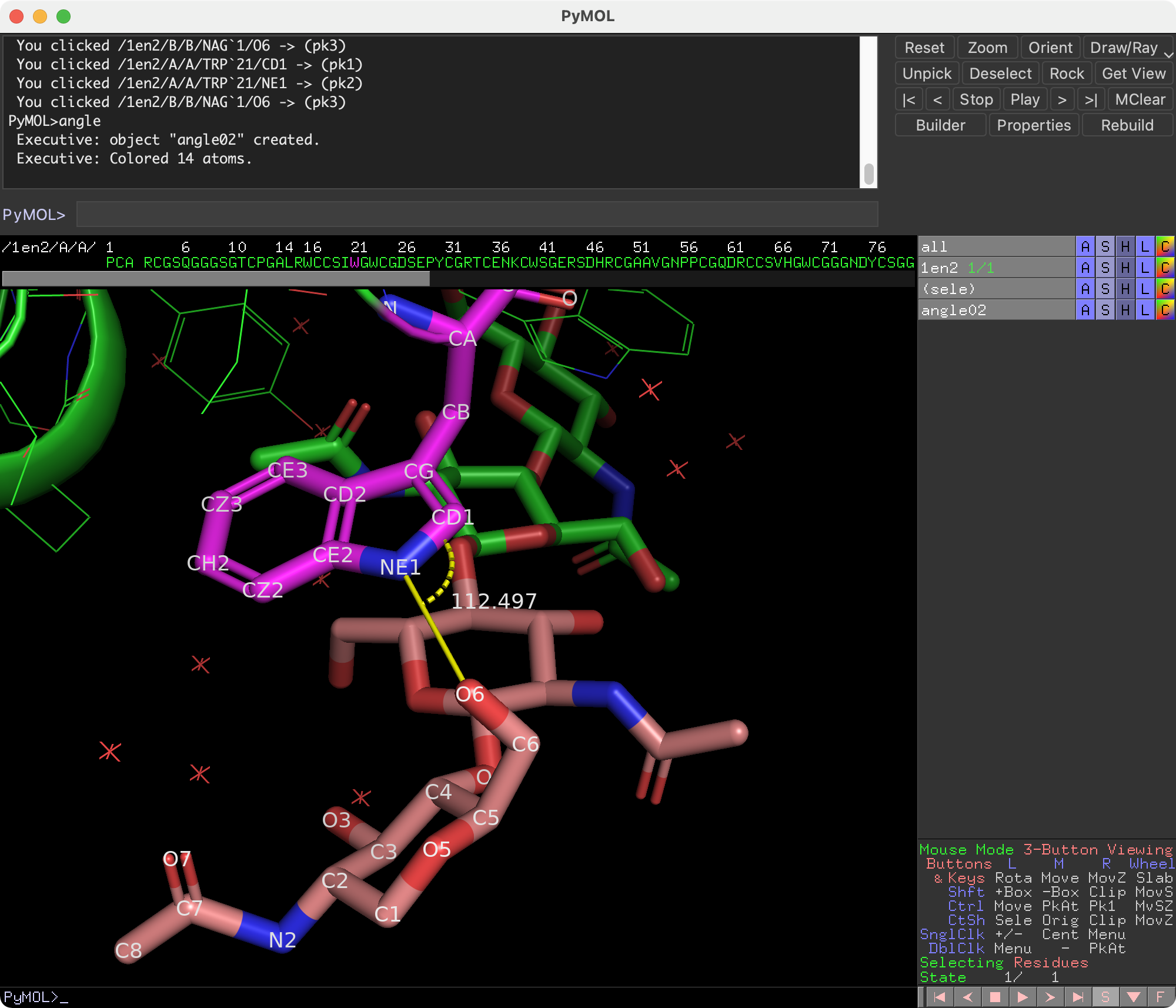
この角度の値112.497°を取得してみます。例1のときの#%%までは同様です。
角度の計算は、Bio.PDB.Vectorモジュールに含まれる関数calc_angle(vector1, vector2, vector3)で行うことができます。各パラメータのvectorには、atomオブジェクトについてのメソッド.get_vector()を適用してベクトル化しておいた座標情報を入力します。
calc_angle()で得られる角度はradian単位になっています。標準ライブラリmathのメソッドdegrees()を使うことでdegree単位に変換することができます。
atomAvec = struc[0]["A"][21]["CD1"].get_vector()
atomBvec = struc[0]["A"][21]["NE1"].get_vector()
atomCvec = struc[0]["B"][("H_NAG", 1, " ")]["O6"].get_vector()
# angleはradian表記になっている。
angleABC_rad = Bio.PDB.vectors.calc_angle(atomAvec, atomBvec, atomCvec)
print(angleABC_rad) # >>> 1.9634352400286554
# degree表記にしたい場合はmath.degrees()を使う。
import math
print(math.degrees(angleABC_rad)) # >>> 112.49655260089772
例3: 4点の原子が作る二面角(torsion angle/dihedral angle)を計算する
例として1EN2のChain AのTRP-21のCG原子、CD1原子、NE1原子とChain BのNAG-1のO6原子が作る二面角(°)を取得してみます。PyMOLで表示するとこんな感じです。
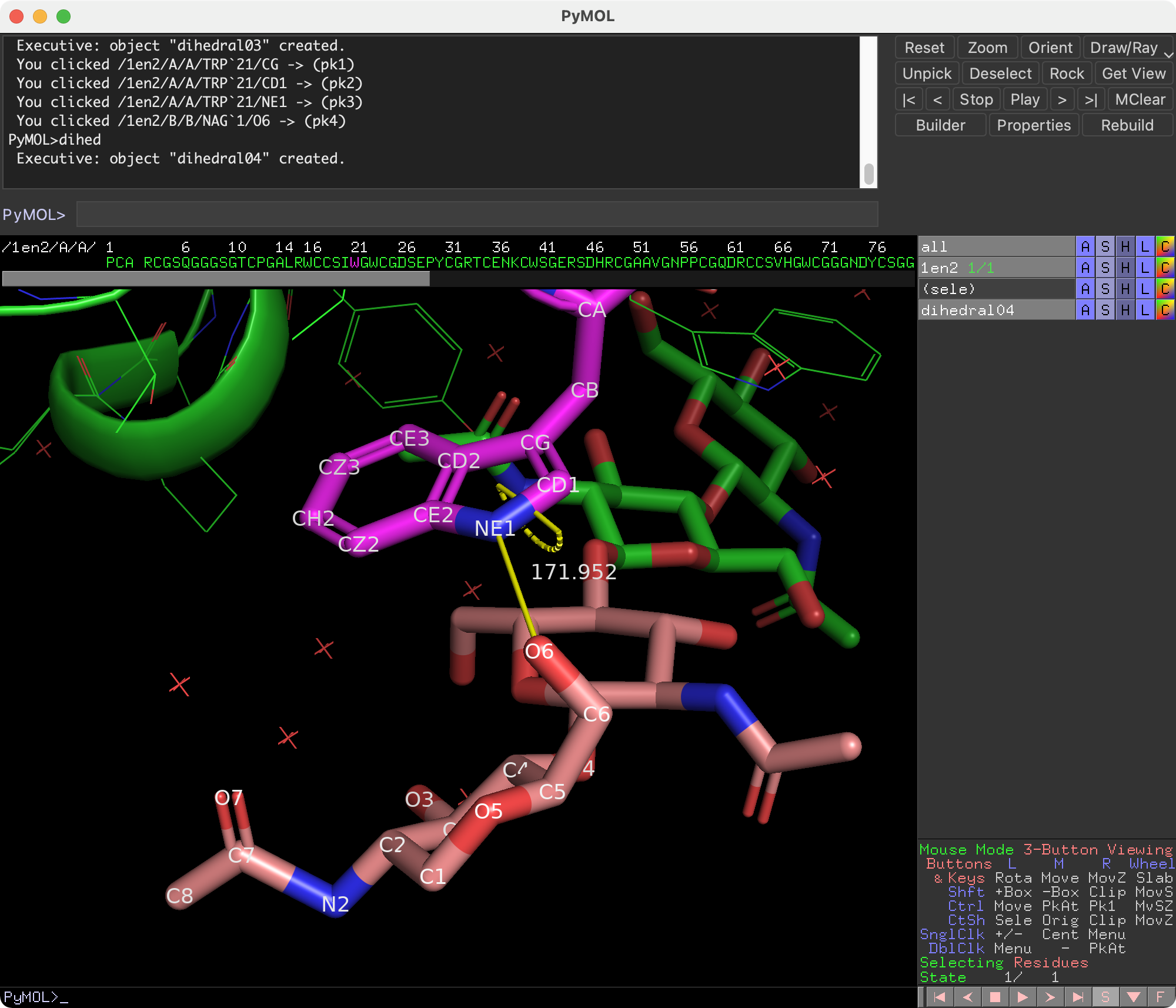
化合物やタンパク質において、原子がA-B-C-Dと結合しているときの二面角が、化合物・タンパク質の立体配座を決定する要素のひとつとして重要です。これは二面角は結合距離や結合角に比べ自由度が大きいためです。
二面角は距離や角度に比べると測定方法が少しわかりにくいですが、PyMOL上のようなViewer上では以下のように考えると簡単です。
- Viewer上で2番めの原子Bと3番めの原子Cを、Bが手前、Cが奥に重なるように視点を動かします。
- このときのA-(B/C)-Dの形成する角度が二面角です。
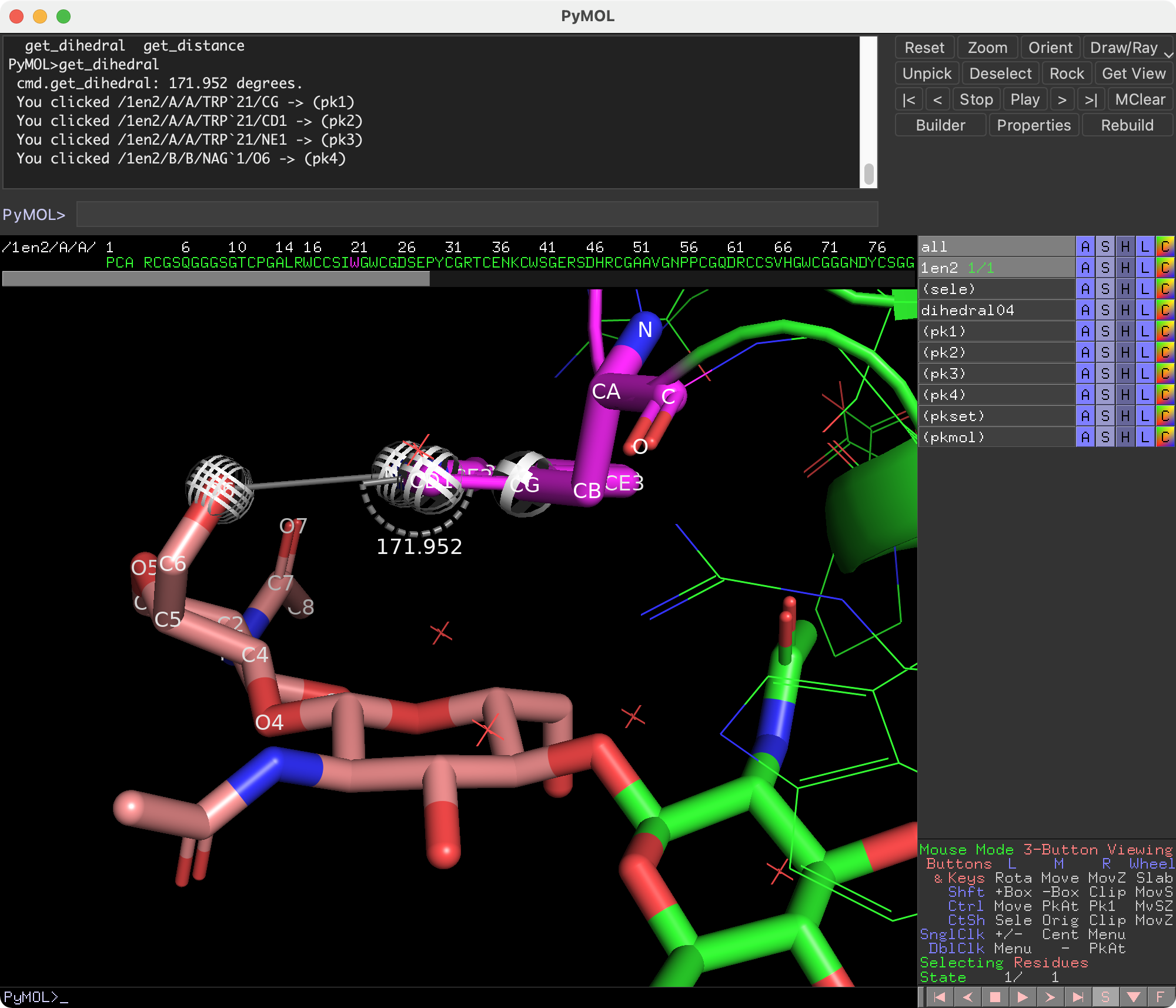
上記1,2の操作を行った時、A-BとC-Dのなす面が時計回り方向である方を正とします。反時計回りにすると負の角度になります。つまり、ある二面角が$-30 ^\circ$であれば、$330 ^\circ$と表すこともできます。もっと一般的な書き方をすれば$n$を整数として$(-30 \pm 360n) ^\circ$となります。
atomAvec = struc[0]["A"][21]["CG"].get_vector()
atomBvec = struc[0]["A"][21]["CD1"].get_vector()
atomCvec = struc[0]["A"][21]["NE1"].get_vector()
atomDvec = struc[0]["B"][("H_NAG", 1, " ")]["O6"].get_vector()
# angleはradian表記になっている
dihedABCD_rad = Bio.PDB.vectors.calc_dihedral(atomAvec, atomBvec, atomCvec, atomDvec)
print(dihedABCD_rad) # >>> 3.0011356947269507
import math
print(math.degrees(dihedABCD_rad)) # >>> 171.9524090539165
NeighborSearchモジュールを使った近傍探索
NeighborSearchモジュールは以下の2つの目的に有用なので紹介しておきます。
- 与えられた位置座標から一定の距離以内に存在するすべてのStructure/Model/Chain/Residue/Atomを検索する
- 互いにある一定の距離以内に存在するすべてのStructure/Model/Chain/Residue/Atomを検索する
1.の目的ではsearch()メソッドを使います。2. の目的ではsearch_all()メソッドを用います。
NeighborSearchクラスのインスタンスを生成するときに引数として原子のリストatom_listを渡す必要があります。これが上記1. 2.の目的での検索対象の範囲になります。
例として、PDB ID: 1ALKのChain Aの残基番号85の残基を構成するすべての原子から5Å以内に存在する原子をすべて取得するコードを示します。
import gzip
import urllib.request
from mimetypes import guess_type
import Bio.PDB
from Bio.PDB.MMCIFParser import FastMMCIFParser
from Bio.PDB.NeighborSearch import NeighborSearch
def _open(file):
encoding = guess_type(file)[1]
if encoding == "gzip":
return gzip.open(file, mode="rt")
else:
return open(file)
urllib.request.urlretrieve("https://files.rcsb.org/download/1alk.cif.gz", "1alk.cif.gz")
# %%
ciffile = "1alk.cif.gz"
pdb_parser = FastMMCIFParser(QUIET=True)
with _open(ciffile) as handle:
struc = pdb_parser.get_structure("1alk", handle)
# %%
"""
NeighborSearchでの検索範囲を設定
Bio.PDB.Selection.unfold_entities()を使って"A"tomオブジェクトのリストを取得
"""
atom_list = Bio.PDB.Selection.unfold_entities(struc, "A")
# 中心となる残基の設定。Chain Aの残基番号85を指定。
target_residue = struc[0]["A"][85]
# NeighborSearchのインスタンスを設定。
ns = NeighborSearch(atom_list)
"""
set()は集合型であることを表す。集合型は重複を自動的に除去する。
https://qiita.com/Tocyuki/items/0bc783daab382ef7a0ec
nearby_atomsに、target_residueを構成する原子から5Å以内に存在する
すべての原子が重複なく取得される
"""
nearby_atoms = set()
for target_atom in target_residue:
nearby_atoms = nearby_atoms.union(ns.search(target_atom.get_coord(), 5, "A"))
"""
上の3行を省略して1行で書くやり方もある(高度)
参考:多重ループを使ったリストの作成をリスト内包表記で行う(https://www.javadrive.jp/python/list/index17.html)
"""
# nearby_atoms = set(atom for target_atom in target_residue for atom in ns.search(target_atom.get_coord(), 5, "A"))
# nearby_atomsの個数を表示
print(len(nearby_atoms)) # >>> 57
このコードが正しく動いているかを、PyMOLの機能と比較して調べてみましょう。PyMOLで1ALKの構造を開き、以下のPyMOLコマンドを入力します。
# Chain Aかつ残基番号85を選択
select resi 85 and chain A
# 選択している範囲`sele`から5Åのすべての残基(resn *)を選択
select resn * within 5 of sele
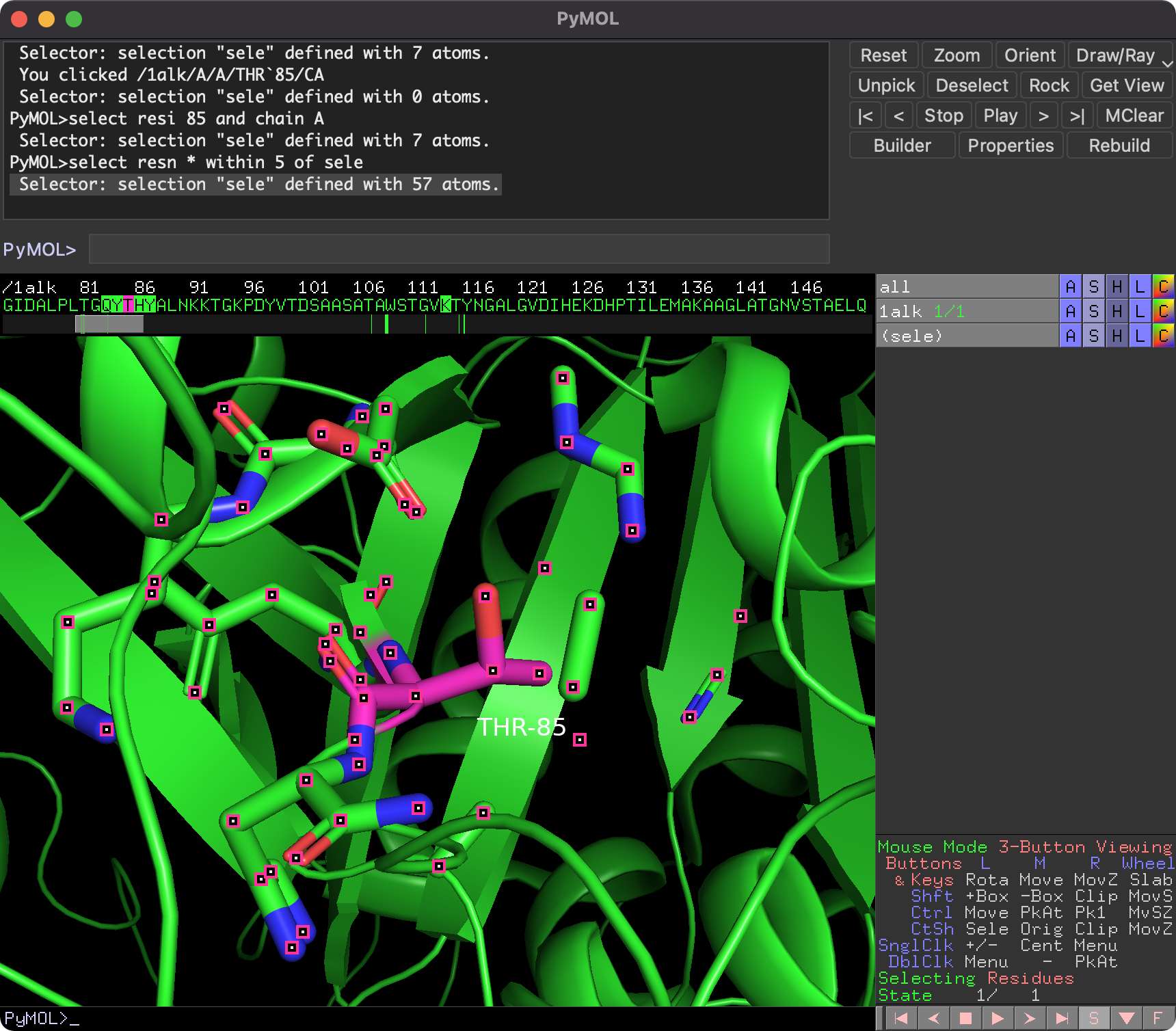
結果、selection "sele" defined with 57 atomsと表示されているように、求める原子の数は57でした。これは上のスクリプトの結果と一致しています。
ちなみに上のコードの例ではDisorderedResidue/Atomについては考慮していません。考慮する必要がある場合は、atom_listにDisorderedAtomについても追加する必要があります(Bio.PDB.Selection.unfold_entities(struc, "A")では入っていない)。
NeighborSearchモジュールは速さ重視のためにC言語で実装されたKD treeを利用しています。
参考:
- https://biopython.org/docs/1.75/api/Bio.PDB.NeighborSearch.html
- https://biopython.org/docs/1.75/api/Bio.PDB.Selection.html
構造情報のPDB/mmCIFフォーマットへの書き出し
変数に格納された構造情報は、MMCIFIOクラスまたはPDBIOクラスを使うことで、それぞれmmCIFフォーマットまたはPDFフォーマットで書き出すことができます。
import gzip
import urllib.request
from mimetypes import guess_type
import Bio.PDB
from Bio.PDB.mmcifio import MMCIFIO
from Bio.PDB.MMCIFParser import FastMMCIFParser
from Bio.PDB.PDBIO import PDBIO
def _open(file):
encoding = guess_type(file)[1]
if encoding == "gzip":
return gzip.open(file, mode="rt")
else:
return open(file)
urllib.request.urlretrieve("https://files.rcsb.org/download/1en2.cif.gz", "1en2.cif.gz")
ciffile = "1en2.cif.gz"
pdb_parser = FastMMCIFParser(QUIET=True)
with _open(ciffile) as handle:
struc = pdb_parser.get_structure("1EN2", handle)
# strucをmmCIFフォーマットで書き出す
# ioという名前でMMCIFIOのインスタンスを生成
io = MMCIFIO()
io.set_structure(struc)
io.save("1en2_output.cif")
# strucをPDBフォーマットで書き出す
# io2という名前でPDBIOのインスタンスを生成
io2 = PDBIO()
io2.set_structure(struc)
io2.save("1en2_output.pdb")
これによって、strucに格納された1en2の構造情報を1en2_output.cifまたは1en2_output.pdbに書き出すことができます。この出力ファイルはPyMOLで開いて構造を表示させることもできます。ただし、インプットとした構造情報とアウトプットされたファイルは完全に同一ではなく、ヘッダー情報がなくなっていたり、場合によってはチェイン情報が変化していたりしていますので(バグ?)、取り扱いには気をつけてください。
場合によってはBioPythonを使って取り込んだ構造情報の一部だけを出力したいという場合もあると思います。その例についても紹介します。
構造の一部を書き出したい場合は、PDBIOの下にあるSelectクラス(Bio.PDB.PDBIO.Select)を利用します。Selectには階層ごとに4つのメソッドがあります。
accept_model(model)accept_chain(chain)accept_residue(residue)accept_atom(atom)
デフォルトでは、上記すべてのメソッドは1(またはTrue)を返します(これは、Model/Chain/Residue/Atomが出力に含まれることを意味します)。Selectをサブクラス化(ここではSelectクラスを継承した子のクラスを作ること)し、必要に応じて0(またはFalse)を返すよう指定するで、ModelやChainなどを出力から除外することができます。ちょっと面倒かもしれませんが、慣れてくると非常に強力です。例として次のコードは、グリシン残基のみを出力します。
# クラスGlySelectを定義する。Selectクラスを継承(inherit)している。
class GlySelect(Select):
def accept_residue(self, residue):
# residueの名前を取得し、"GLY"に一致した時、Trueを返す。つまり、出力に含める。
if residue.get_resname()=="GLY":
return True
# GLYでない場合、Falseを返し、出力に含めない。
else:
return False
# 書き出し準備
io = PDBIO()
io.set_structure(struc)
# io.save()の第2引数にGlyselect()を渡すことで、Glyのみの残基を出力対象にする。
io.save("gly_only.pdb", GlySelect())
これはPythonの仕様の1つである クラスの継承(inheritance) を利用しています。簡単に言うと、すでに存在しているSelectクラスの仕様やaccept_residue()メソッドを上書き(オーバーライド)することで書き換えています。ここで、GlySelectクラスから見て継承元のSelectクラスは親クラス、またはスーパークラスと呼びます。
つまり、Selectクラスを継承した適当な名前のクラスfooを定義し、その中で出力させたい対象についてはreturn Trueを、そうでないものはすべてreturn Falseにしておいて、そのクラスfooをio.save()メソッドで書き出すという流れになります。
以下で2つの例を見てみましょう。
import gzip
import urllib.request
from mimetypes import guess_type
import Bio.PDB
from Bio.PDB.mmcifio import MMCIFIO
from Bio.PDB.MMCIFParser import FastMMCIFParser
from Bio.PDB.PDBIO import PDBIO, Select
def _open(file):
encoding = guess_type(file)[1]
if encoding == "gzip":
return gzip.open(file, mode="rt")
else:
return open(file)
urllib.request.urlretrieve("https://files.rcsb.org/download/1en2.cif.gz", "1en2.cif.gz")
ciffile = "1en2.cif.gz"
pdb_parser = FastMMCIFParser(QUIET=True)
with _open(ciffile) as handle:
struc = pdb_parser.get_structure("1EN2", handle)
# %%
# 例1:PDB ID:1EN2の構造を読み込み、その中からGly残基のみのPDBファイルを書き出す
class ResSelect1(Select):
def accept_residue(self, residue):
# residueの名前を取得し、"GLY"に一致した時、Trueを返す。つまり、出力に含める。
if residue.get_resname() == "GLY":
return True
# GLYでない場合、Falseを返し、出力に含めない。
else:
return False
io = PDBIO()
io.set_structure(struc)
io.save("gly_only.pdb", ResSelect1())
# %%
# 例2:PDB ID:1EN2の構造を読み込み、その中から残基番号21-40の範囲の残基のみのPDBファイルを書き出す
class Resselect2(Select):
def accept_residue(self, residue):
"""
residue.get_id()で残基のIDを取得する。
ただし、residue.get_id()だけだと、例によって('H_PCA', 1, ' ')のような3つ組記法で得られるので
2番めの数値のみを取り出すために[1]をつける必要がある。
"""
if start_resnumber <= residue.get_id()[1] <= end_resnumber:
return True
else:
return False
# はじまりの残基番号を21にセット
start_resnumber = 21
# 終わりの残基番号を40にセット
end_resnumber = 40
io = PDBIO()
io.set_structure(struc)
io.save("Residues21_40.pdb", Resselect2())
参考