AmberToolsに実装されているモジュールantechamberを使って、分子動力学シミュレーション用にあるリガンド小分子の電荷パラメータを決定してみます。
MDシミュレーションを行うとき、ふつうの20種類のアミノ酸や一部の核酸など、よく登場する分子については力場パラメータおよび電荷が設定されていることが多く、これらを簡単に利用することが可能です。しかし、そうは言っても、蛋白質は様々な小分子と結合して化学反応(いわゆる酵素反応)を行うことのがほとんどです。そのため、自分の標的とする蛋白質が結合する小分子の挙動も含めてシミュレーションしたい場合には、自分でパラメータを作ってやる必要があります。
小分子の設定ファイルを作る場合、基本的に必要なのは力場パラメータと電荷パラメータの2つです。力場パラメータとは、それぞれの原子間に存在する化学結合にかかる力のパラメータです。通常、この力のパラメータは2原子間(平衡結合長)だけではなく、つながる3原子間(平衡角度)、4原子間(平衡二面角)の組み合わせを規定する必要があります。
例えばエチレンC2H4だと、平衡結合長の規定はC-H結合とC=C結合の2種類で済みますが、H-C-Hの平衡角度が120度になるために力を設定してやったり、H-C-C-Hの平衡二面角が0度(つまり平面分子)になるよう設定してやる必要があるわけです。
これだけ聞くとすべてを的確に設定してやるのは非常に面倒そうですが、AMBERが提供する汎用力場GAFFを用いると大抵の場合は自動で生成してくれます。原子がH, C, N, O, P, Sとハロゲンから構成される分子にしか適用できない(というかそれ以外の原子相手だと一般化できないため量子計算する必要が発生する)のですが、だいたいの生体分子はこれらでOKです。
続いて、電荷パラメータですが、これは個別に1つ1つ計算する必要があるのが厄介です。例えばメタンCH4のC原子はほぼ中性ですが、カルボニル基C=Oを持つような分子のCはやや+になっているということは、化学をやったことがあるひとなら知っていると思います。 現在のところ、この電荷パラメータを設定する上で最もよく使われているのはRESP法と呼ばれる手法です。これは量子計算で得られた静電ポテンシャルにフィットするようMD用に変換した形式電荷で、RESP電荷と呼ばれます。この記事では、目的の分子のRESP電荷を求める方法について述べます。
環境
- macOS または Linux環境。
- AmberTools20以上がインストールされている
- Gaussian 16がインストールされている
- GaussView 6がインストールされている(Optional), ない場合はAvogadroというフリーソフトで、一応構造を確認することはできます。
商用ソフトウェアのGaussianの代わりに、もしかすると(ユーザー登録すれば)フリーで使えるGAMESSでもできるかもしれません。が、ここではGaussianでのやり方を説明します。GaussViewは単にインプットファイルの作成を簡単にするためのツールなので必須というわけではありません。
今回はDigoxigeninという化合物についてのパラメータを作成する方法を例にとって説明します。
Step 1. GaussView 6での構造計算セットアップ
DigoxigeninはPubChemに化合物として登録されています。このウェブサイトの"3D Conformer"の"Download"にSDF形式での配布が行われているので、SDF SAVEを押して構造データをダウンロードします。もし、Pubchemなどに化合物データが無い場合でもGaussViewのGUIで化合物の骨格をなんとなく作ることはできますので、任意の化合物について計算をしたい場合にはこちらを利用してください。
次にGaussView 6を開きます。File -> Openから先程のSDFファイルを選択すると、GaussView上に構造が表示されます。Mac OSのGaussViewの場合、化合物画面(紫色背景)の上で、Command + Gを押すと"Gaussian Calculation Setup"の画面が表示されます。左下の'submit'→'save'を選択してFilenameをdigoxigenin.gjfとして保存します。保存後"Submit the following file to Gaussian?"と尋ねられるかもしれませんが、これはNoで構いません(Yesにすると手持ちのマシンのGaussianで構造最適化計算を始めようとする)。

続いて、いま保存したdigoxigenin.gjfのファイルをテキストエディタで開きます。このようになっているはず。
%chk=digoxigenin.chk
# hf/3-21g geom=connectivity
15478
0 1
O -1.55630000 1.51150000 0.56430000
O -0.77370000 -3.25260000 -0.03220000
(以下座標データ)
H -5.19080000 -1.90660000 -0.61770000
H -4.51910000 -1.70010000 1.02530000
H -3.64260000 1.84010000 -0.81030000
H 6.95950000 -0.23490000 -1.03450000(ここまで座標データ)
1 6 1.0 57 1.0
2 11 1.0 58 1.0
3 24 1.0 62 1.0
4 26 1.0 28 1.0
5 28 2.0
6 7 1.0 8 1.0 15 1.0
(以下結合情報パラメータ)
今回は構造最適化の計算レベルをB3LYP/6-31+g(d,p)にしたいということや、構造最適化計算を指定すること(opt)、他の計算上の様々な設定を最初のセクションに書く必要があります。ここについての詳細はGaussian 16のgjfファイルフォーマット形式の解説ページを参照されたし。そこでここの上の部分を以下のように変更します。
%chk=digoxigenin.chk
%mem=60GB
%nprocshared=16
#p opt=(tight) scf=qc b3lyp/6-31+g(d,p)
Digoxigenin Downloaded from https://pubchem.ncbi.nlm.nih.gov/compound/15478
0 1
O -1.55630000 1.51150000 0.56430000
O -0.77370000 -3.25260000 -0.03220000
(以下座標データ)
次にGaussian 16を計算させるマシンとの兼ね合いで、%mem=60GBは計算ノードの最大メモリ数の9~9.5割くらいの数字を(10割にはしない方がいいらしい)、%nprocshared=16は計算ノードのCPU数を指定します。Digoxigenin Downloaded~~の部分はコメント行なので何を書いても良い。
geom=connectivityのオプション設定をここでは消しています。このオプションは今回のような純粋な量子化学計算をする場合には不必要である。ただしGaussian calculation set upのときにはこのオプションがデフォルトで指定されていることが多い(チェックを外せばいいだけなんだけど)。このオプションが入っていると結合情報パラメータも明示する必要があり、逆にオプションがない場合には結合情報パラメータは消す必要があります。
Gaussianでは空行はセクションの終了を規定する意味を持つので、セクション区切りとしての適切な位置での空行を忘れてはいけません。また最後に1行だけ空行を挟むことも要求されます。
これでセットアップ完了。
Step 2. Gaussian 16での構造最適化
この作成したdigoxigenin.gjfファイルをGaussian 16が載っている計算機(LinuxのCentOS 7が良い)で計算させます。
スパコンでqsubで計算を投入させるときのシェルスクリプトはこんな感じ(PBS系のキューイングシステムのqsubの例です)。
#!/bin/sh
#PBS -l select=1:ncpus=16,walltime=72:00:00
#PBS -q default
test $PBS_O_WORKDIR && cd $PBS_O_WORKDIR
job="digoxigenin"
g16 < ${job}.gjf > ${job}.log
最後のjob="digoxigenin" ; g16 < ${job}.gjf > ${job}.logの部分が計算部分の本体です。それより前の部分は計算機のqsub環境に合わせて変更する必要があります。詳しくは各計算機リソースの説明ページを読んでね。
構造最適化計算が始まるとdigoxigenin.chkやdigoxigenin.logが書き込まれます。もし計算が始まって数分で終了している場合、空行を挟み忘れたとかgeom=connectivityと結合情報のミスマッチとかが考えられます。
私がやってみたところ、計算の正常終了までにだいたい3〜4時間ほど必要でした。計算途中に構造最適化によってエネルギーが落ちていく様子はこのログファイル内のSCF Done:をサーチする次のコマンドで見られます。
$ grep "SCF Done" digoxigenin.log
SCF Done: E(RB3LYP) = -1273.00574261 a.u. after 8 cycles
SCF Done: E(RB3LYP) = -1273.01731710 a.u. after 11 cycles
SCF Done: E(RB3LYP) = -1273.01926851 a.u. after 11 cycles
SCF Done: E(RB3LYP) = -1273.01990688 a.u. after 11 cycles
SCF Done: E(RB3LYP) = -1273.02028583 a.u. after 11 cycles
SCF Done: E(RB3LYP) = -1273.02055300 a.u. after 11 cycles
SCF Done: E(RB3LYP) = -1273.02073084 a.u. after 9 cycles
エネルギーがどんどん低くなっていれば良い感じ。また、qsubジョブが終了してるときに、正常終了したか異常終了したかを必ず確認する必要があります。正常終了している場合にはこのログファイルの最後が
Job cpu time: 1 days 10 hours 47 minutes 37.6 seconds.
Elapsed time: 0 days 2 hours 54 minutes 18.0 seconds.
File lengths (MBytes): RWF= 124 Int= 0 D2E= 0 Chk= 20 Scr= 1
Normal termination of Gaussian 16 at Mon Mar 26 16:53:35 2018.
のようにNormal terminationと表示される。逆に異常終了している場合はError terminationのはず。構造最適化計算の途中で異常終了することはまあまあよくあります。そんなときはGaussian計算の"Restart"をしかけたり、構造最適化計算の最後の構造を抜き出してもう一度gjfインプットファイルを作成し直して再投入するなどの方法が考えられます。ここについては詳しい人に尋ねてみてください。
無事Gaussianのログファイルの最終行がNormal Terminationと表示されて計算が正常終了することを迎えられたら次のステップに行きます。
Step 3. Gaussian 16で計算レベルを変え、iopオプションを付けて計算
構造最適化されたdigoxigenin立体構造が得られたら、それをもとにもう一度だけ追加計算をします。今度はこの構造最適化された立体構造を変えないまま量子計算の計算レベルをHF/6-31g(d)に、さらにdigoxigeninの各原子について量子計算の結果からタンパク質力場に沿った点電荷パラメータを計算して生成するという作業を行います。
先程の構造最適化されたログファイルをMacのGaussViewで開き、最後の構造を表示させた状態でCommand + G (Mac)またはCtrl+G (Win)を押してGaussian Calculation set upを表示させ、その座標のgjfファイルを作成する。Filenameはdigoxigenin_resp.gjfとします。このファイルをテキストエディタで開き、今度は以下のようにセクションを書き換えます。
%chk=digoxigenin_resp.chk
%mem=60GB
%nprocshared=16
#p hf/6-31g(d) pop=mk iop(6/33=2,6/42=6) scf=tight
Optimized digoxigenin structure
0 1
O 1.41385500 -1.92103000 0.34991100
O 1.03551400 2.98380900 0.02168900
(以降構造最適化されたあとの座標データ)
ここではhf/6-31g(d) pop=mk iop(6/33=2,6/42=6)となっていること、geom=connectivityとoptを指定していないことが重要です。これを作り、再びGaussian 16でこのdigoxigenin_resp.gjfを計算させます。およそ20〜30分程度で終わります。
Step 4. antechamberモジュールでのRESP電荷変換
このdigoxigenin_resp.logが正常に計算終了していることを確認したら、AMBER22 (AmberTools22)に同梱されているantechamberを使うと、RESP電荷の含まれる構造ファイルに変換することが簡単にできます。MacにもAMBER22をインストール可能です。antechamberの絶対PATHは、AMBERのインストールディレクトリを$AMBERHOMEとしたときの$AMBERHOME/bin/antechamberとなっているはずです。
コマンドは以下の通り。antechamber -hでヘルプを表示できます。
antechamber -i digoxigenin_resp.log -fi gout -o dig.mol2 -fo mol2 -c resp -at gaff2 -nc 0 -m 1 -rn DIG -pf y
-iはインプットファイル名、-fiはインプットファイルタイプの指定、-oはアウトプットファイル名の指定、-foはアウトプットファイルタイプの指定。
これら4つは必ず指定しておく必要があります。ファイルタイプの対応一覧やヘルプantechamber -hで確認可能。
-at gaff2はmol2形式ファイルの原子種(atom type)の設定をgaff2方式で行うというものです。gaff2の他にはsybylとかgaff(ふるいバージョン)などがあります。
-c respはRESP電荷の割当。Gaussian 16のインプットファイル時に適切に指定しておく必要があります。他にはam1-bccなどがあります。
-nc 0 -m 1net charge(系全体の電荷)とmultiplicity(スピン多重度)の設定。Gaussian
16の入力と同じものにしておきます。
-nc 0 -m 1は化合物ごとに変わるので注意しましょう。
-rn DIG出力される残基名をDIGとします。後に手動で容易に変更できるので付け忘れても良い。
-rn DIGはリガンドの3文字表記をつけているだけなので、DIG以外でも好きに付けてOKです。自分にとって馴染みのある名前にしておきましょう。
-pf yを付けておけば終了時に中間ファイルが削除されるようになります。
SYBYLのAtom Typeについてはこちらのページを参照。Aromaticな炭素・窒素原子はarと書き直した方がよいのかもしれませんが、ここではこのままにしておきます。。
うまく行っているときの表示例
$ antechamber -i digoxigenin_resp.log -fi gout -o dig.mol2 -fo mol2 -c resp -at gaff2 -nc 0 -m 1 -rn DIG -pf y
Welcome to antechamber 22.0: molecular input file processor.
acdoctor mode is on: check and diagnosis problems in the input file.
-- Check Format for Gaussian Output File --
Status: pass
-- Check Unusual Elements --
Status: pass
-- Check Open Valences --
Status: pass
-- Check Geometry --
for those bonded
for those not bonded
Status: pass
-- Check Weird Bonds --
Status: pass
-- Check Number of Units --
Status: pass
acdoctor mode has completed checking the input file.
WARNINGはまだしもErrorが出た場合にはメッセージを良く読んで対処してみてください。エラーが表示されていなければ、とりあえずこれでRESP電荷の計算が終わります。
Step 5. RESP電荷の細かな調整(optional)
このできあがったdig.mol2ファイルをテキストエディタで開いてみると次のようになっているはずです。
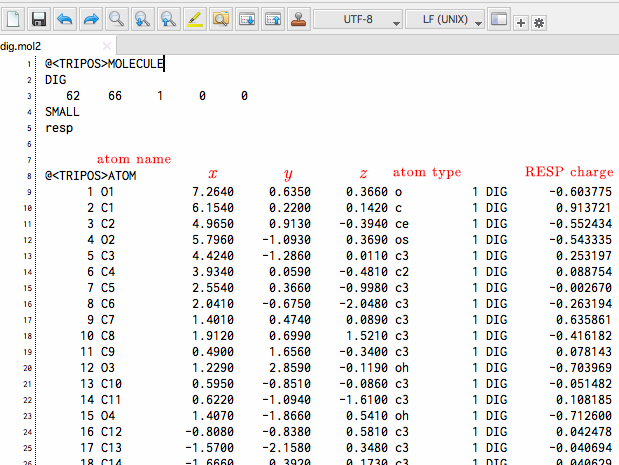
このうちRESP chargeを表すカラムは右端の数字ですが、AMBERに読み込ませる場合は色々あって小数点以下5桁までで調整していたほうが都合がよいので、なんらかの方法を使ってここの数字を小数点以下5桁にしておいてください。最も簡単な方法はこのファイルをExcelで開いて適当に四捨五入です。また、このRESP chargeの合計値を小数点以下.000000にしておく必要があります。antechamberで変換した直後では、往々にして.999999とか, .000001とかになっていることがありますので、そこは適宜調節してやってください。
以下変換した時の例
@<TRIPOS>MOLECULE
DIG
62 66 1 0 0
SMALL
resp
@<TRIPOS>ATOM
1 O1 7.2640 0.6350 0.3660 o 1 DIG -0.603790
2 C1 6.1540 0.2200 0.1420 c 1 DIG 0.913720
3 C2 4.9650 0.9130 -0.3940 ce 1 DIG -0.552430
4 O2 5.7960 -1.0930 0.3690 os 1 DIG -0.543340
5 C3 4.4240 -1.2860 0.0110 c3 1 DIG 0.253200
(以下省略)
この例では全体で.000000になるように、1行目のO1のRESP電荷を-0.603775から調節して-0.603790にしています。
Step 6. parmchk2で足りない力場パラメータを追加する(optional)
今回の小分子パラメータの例では必要ないのですが、分子によっては追加の力場パラメータを生成する必要があります。
/Users/YoshitakaM/apps/amber22/bin/teLeap: Error!
** No torsion terms for atom types: ca-ca-ce-ha
そんなときはparmchk2コマンドで足りていない力場パラメータを作り出すことができます。
parmchk2 -i dig.mol2 -f mol2 -o frcmod.dig -s gaff2
AmberToolsを使ったleapのときにそのファイルを読み込むように追記することで、leapによるMDシミュレーションの準備を行うことができるようになります。
#AMBER の力場パラメータff14SBを読み込む
source leaprc.protein.ff14SB #for AmberTools22
source leaprc.gaff2 #小分子向けの汎用力場「GAFF2」を読み込む
source leaprc.water.tip3p #for AmberTools22
#追加のイオンの力場の導入
loadAmberParams frcmod.ionsjc_tip3p
loadAmberPrep ./frcmod.dig
#.mol2ファイルの読み込み
DOG = loadMol2 ../PDB/dog.mol2
loadAmberPrep ATP.prep
loadAmberPrep Mg.prep