AlphaFold3とalphafold3_toolsで様々なタンパク質-基質複合体予測を行うチュートリアル
GitHubにalphafold3_toolsというツールを作成したので、その宣伝をかねて、これを使ってリガンド情報を追加したAlphaFold3の構造予測を行う方法を紹介します。
Requirements
- AlphaFold3をインストールしてある
- Python3.10以降が動作できる環境(macOSだとHomebrew, Linuxだと最初から入っていると思う)
題材
題材としてMycobacterium tuberculosis(結核菌)由来のタンパク質UniProt ID: P9WIC5のChorismate pyruvate-lyaseの酵素基質複合体を予測してみます。
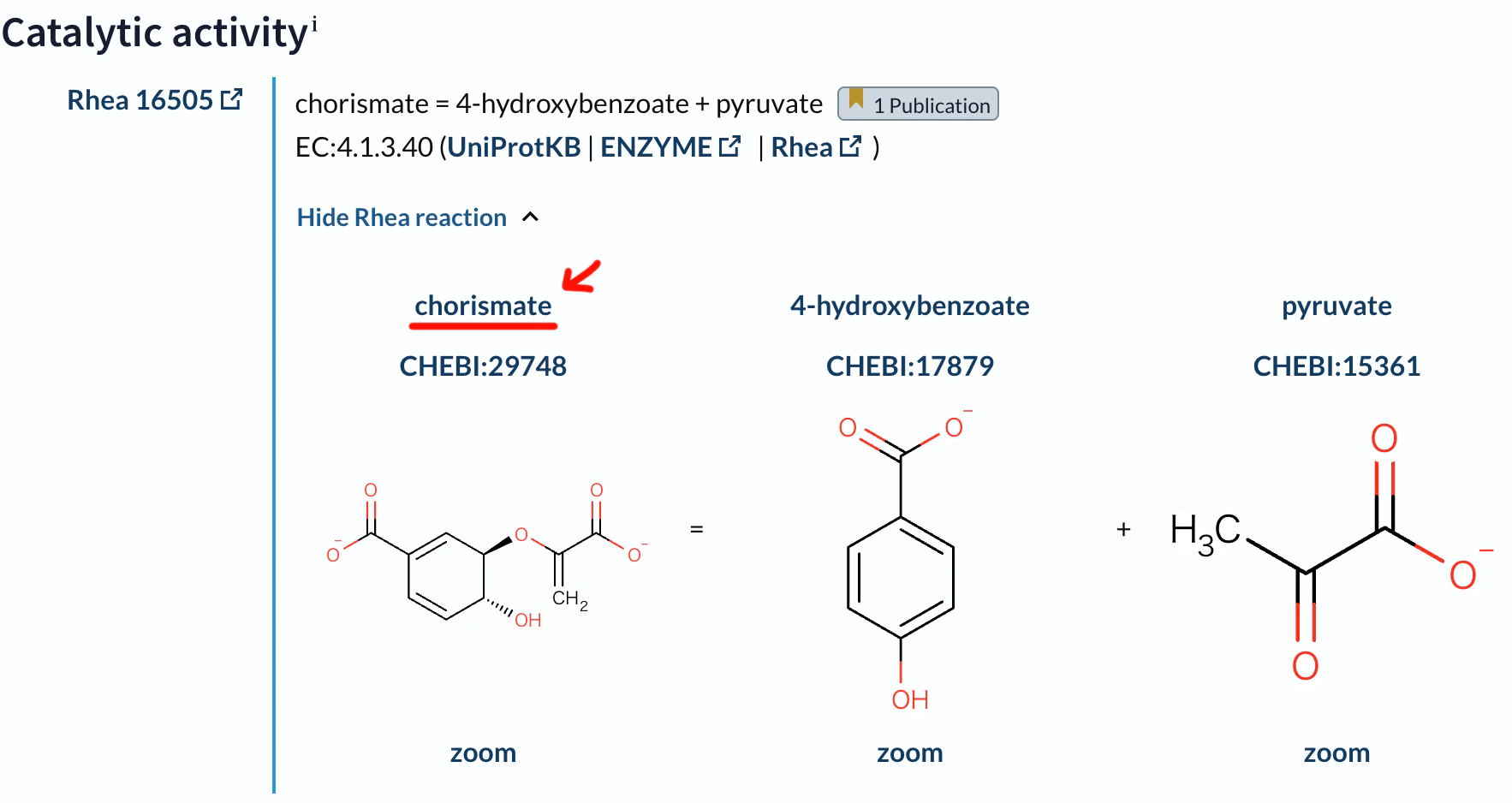
この反応を触媒する酵素です。現時点で結晶構造は決定されていません。
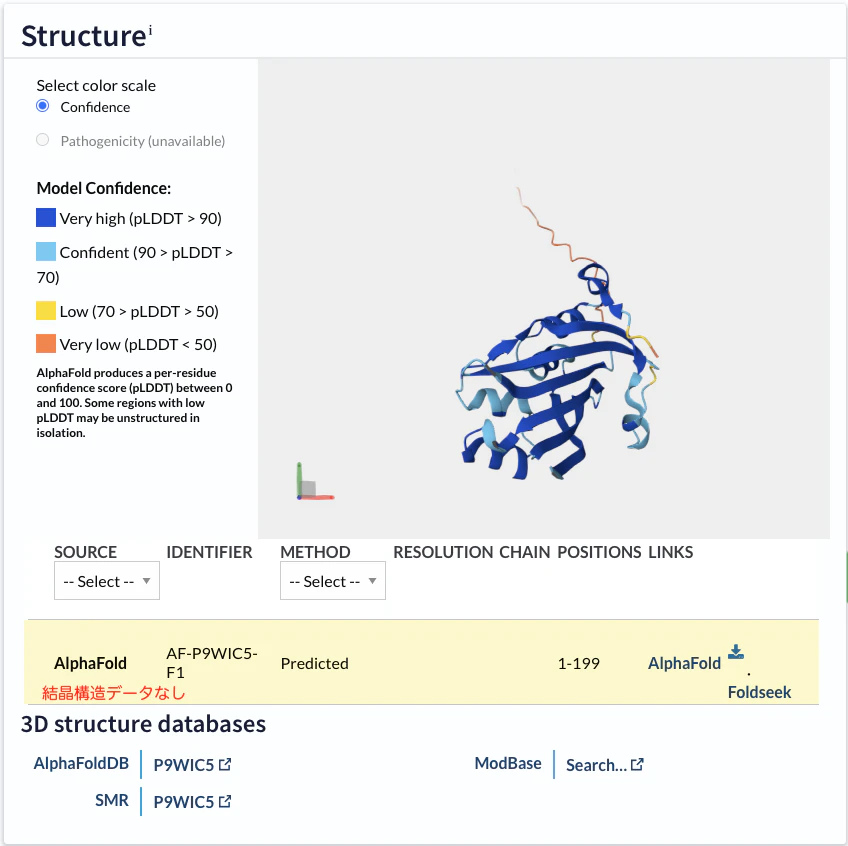
アミノ酸配列は同ページのSequenceのところから取得することができますので、これをAlphaFold3への予測対象配列とします。
>P9WIC5
MTECFLSDQEIRKLNRDLRILIAANGTLTRVLNIVADDEVIVQIVKQRIHDVSPKLSEF
EQLGQVGVGRVLQRYIILKGRNSEHLFVAAESLIAIDRLPAAIITRLTQTNDPLGEVMA
ASHIETFKEEAKVWVGDLPGWLALHGYQNSRKRAVARRYRVISGGQPIMVVTEHFLRSV
FRDAPHEEPDRWQFSNAITLAR
リガンド(基質)はchorismate (コリスミ酸)とします。
MSAを取得する
AlphaFold3は、AlphaFold2時代と同じく、まず予測したいアミノ酸配列の類縁配列であるMultiple Sequence Alignment (MSA)を取得する処理を行ってから、これに潜む情報を加工して、続く構造予測処理へと入っていきます。前半のMSA取得はCPUオンリーの処理であり、後半の構造予測はGPUを使う処理で、原理的には分離して計算させることが可能です。構造予測処理部分は、短い配列(200〜300)のもので1分程度、長い配列(3000〜4000)のもので30〜50分程度です。
MSA取得処理についてはいくつかやり方があるので、それぞれ紹介します。
1. AlphaFold3にそのままお任せする
AlphaFold3はHMMERを使ったMSA取得の機能が備わっているので、これを使うことがstraightforwardです。以前のAlphaFold3インストール記事のテストランのところで、予測配列対象を上記のアミノ酸配列に変えて予測させれば、それだけで自動的にMSAの取得と構造予測が続けて実行されます。
{
"name": "P9WIC5_pred",
"modelSeeds": [1],
"sequences": [
{
"protein": {
"id": ["A"],
"sequence": "MTECFLSDQEIRKLNRDLRILIAANGTLTRVLNIVADDEVIVQIVKQRIHDVSPKLSEFEQLGQVGVGRVLQRYIILKGRNSEHLFVAAESLIAIDRLPAAIITRLTQTNDPLGEVMAASHIETFKEEAKVWVGDLPGWLALHGYQNSRKRAVARRYRVISGGQPIMVVTEHFLRSVFRDAPHEEPDRWQFSNAITLAR"
}
},
{
"ligand": {
"id": ["B"],
"smiles": "O[C@@H]1C=CC(=C[C@H]1OC(=C)C([O-])=O)C([O-])=O"
}
}
],
"dialect": "alphafold3",
"version": 1
}
"smiles": "O[C@@H]1C=CC(=C[C@H]1OC(=C)C([O-])=O)C([O-])=O"は予測したいリガンド・コリスミ酸のSMILES表記です(後述)
構造予測が終わると、アウトプットディレクトリに*_data.jsonというファイルが生成されています。このJSONファイルはMSAの情報を含んでおり、再利用することも可能なので、これを改変すれば(例:リガンド部分だけの情報を書き換えるなど)、再度短時間で複合体予測を実行することができます。
しかし、HMMERを使ったMSA取得は一般に10〜20分以上時間がかかるためスループット性はそこまで高くありません。
なので、ここではそんな当たり前な方法ではなく、もっと大量に高速にMSAを取得したい場合も含めて別の方法を紹介します。
2. ColabFoldを使う
AlphaFold2についてのColabFoldは、構造予測の中でMSAを超高速に取得する処理を含んでいます。これを使うことで、MSAを取得することができます。
以下の画像はColabFoldのスクリーンショットです。"sequence"のところにFASTA形式のファイルを入力して、「ランタイム」メニューから「すべてのセルを実行」を選択すると、構造予測を行うと同時に、MSAのファイルも取得できます。
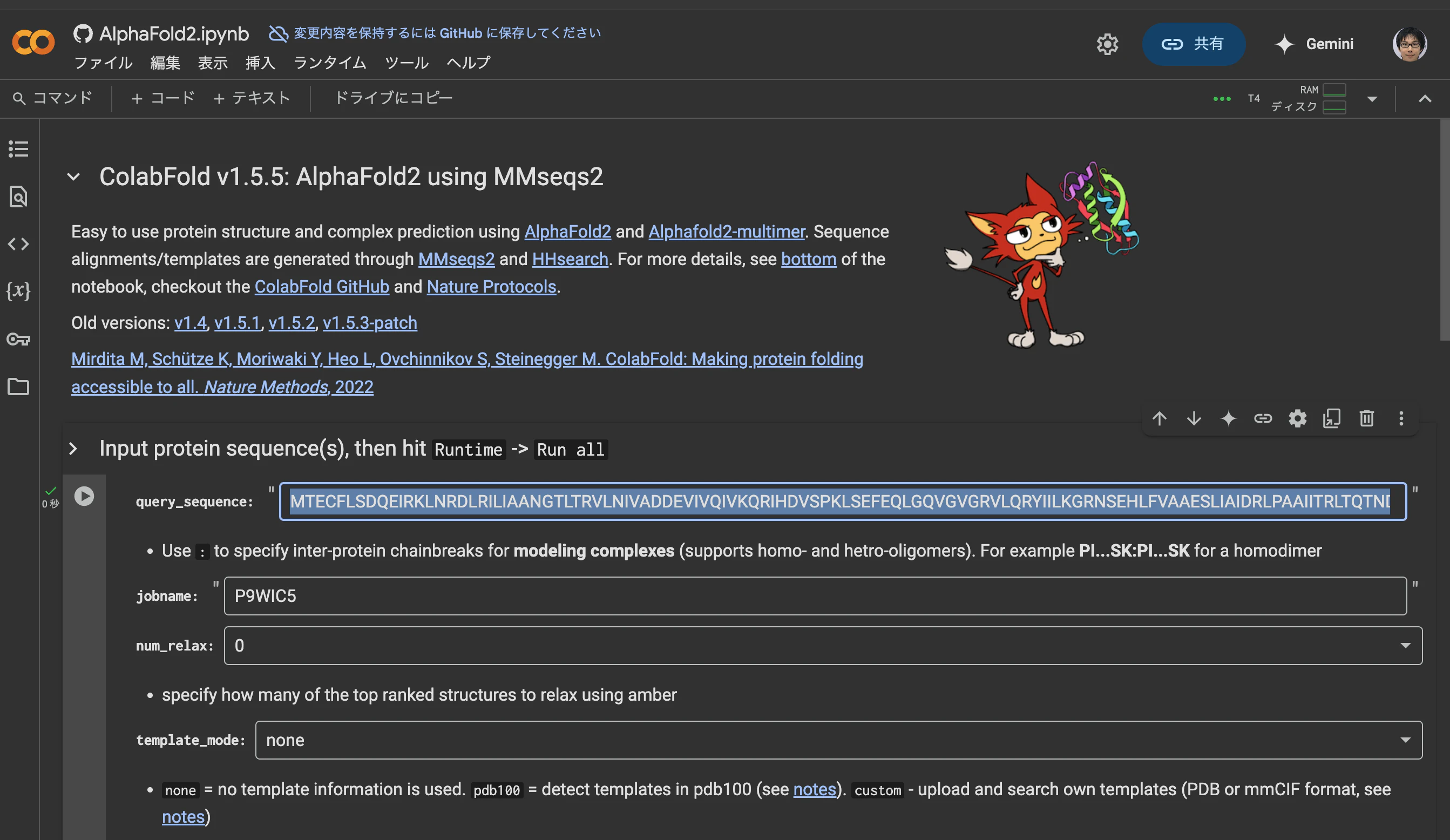
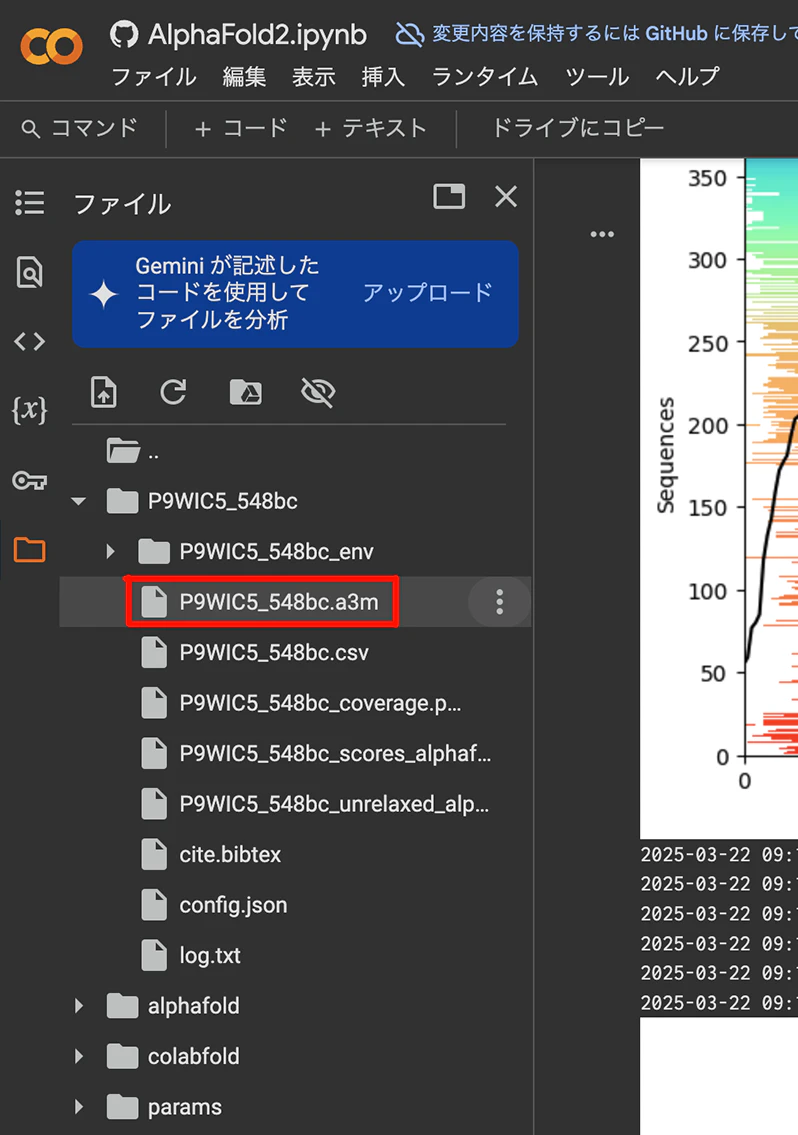
上記画像の赤枠で示された、a3m形式のP9WIC5_548bc.a3mがMSAのファイルとなっています。赤枠の右にあるケバブボタン(︙← こんなボタン)を押すとダウンロードメニューが現れるので、これを押してこちらを手元のPCにダウンロードすればOKです。
_548bcというのは、ColabFoldの実行時に自動生成されるランダムな文字列(ハッシュ値の先頭の文字)です。この文字列は実行ごとに異なります。意味は特にないので気にしなくてOKです。
または、LocalColabFoldでも同様のことができます。その場合は--msa-onlyというオプションをつけて実行することで、MSAのファイルだけを取得することができます。
colabfold_batch --msa-only P9WIC5.fasta outdir
このコマンドで、outdirディレクトリにMSAのファイルP9WIC5.a3mが生成されます。これを後のインプットにします。こちらはハッシュ値はついていません。
outdir
├── P9WIC5.a3m
├── P9WIC5.pickle
├── P9WIC5_coverage.png
├── P9WIC5_env
│ ├── bfd.mgnify30.metaeuk30.smag30.a3m
│ ├── msa.sh
│ ├── out.tar.gz
│ ├── pdb70.m8
│ └── uniref.a3m
├── cite.bibtex
├── config.json
└── log.txt
3. colabfold_searchを使う
LocalColabFoldのcolabfold_searchを使うことで、MSAのファイルを取得することができます。これはColabFoldのMSA取得サーバーとほぼ同じものをローカルで実行するため、最高速度の1配列あたり1分でMSAをを取得できるようにするためにはRAM 768GB以上のマシンが必要となります。詳細は私の以前の記事"LocalColabFoldを完全ローカル環境で動作させる"を参照してください。
名古屋大学のスパコン不老Type-IIIとTSUBAME 4.0にはこの動作のためのデータベースは導入されています。
AlphaFold3のためのJSONファイルの生成
alphafold3_toolsのインストール
GitHubからalphafold3_toolsをインストールします。これは私が作成したソフトウェアで、AlphaFold3のinput JSONファイル作成支援と、アウトプットの可視化を行うことができます。
必要なソフトウェア
- Python 3.10以降
macOSではHomebrewをインストールしておくとbrew install python@3.12でpython3.12をインストールすることができます。最新はpython3.13です。
python3.12以降を使う場合は、まず仮想環境venvを作成してからactivateすることが必須となりました。
/path/to/workingdirectoryは作業ディレクトリです。
mkdir -p /path/to/workingdirectory ; cd /path/to/workingdirectory
python3.12 -m venv .venv
source .venv/bin/activate
# Ubuntu 22.04ではデフォルトでpython3.10が使われます。
# GitHubからインストール
python3 -m pip install git+https://github.com/cddlab/alphafold3_tools.git
# upgrade
python3 -m pip uninstall alphafold3_tools -y && python3 -m pip install --upgrade git+https://github.com/cddlab/alphafold3_tools.git
こののち、ターミナルでmsatojson -hと入力してみてヘルプメッセージが表示されれば成功です。
a3mファイルをJSONファイルに変換
得られたMSAのファイルの中身をAlphaFold3のinput JSONファイル形式に変換します。これにはalphafold3_toolsのmsatojsonというコマンドを使います。
msatojson -i P9WIC5_548bc.a3m -o P9WIC5.json
このインプット-iに与えられるa3mファイルは先程のColabFoldの出力ファイルです。-oには出力ファイル名を指定します。
msatojsonは複数のa3mファイルを含むディレクトリを指定することもできます。この場合、-oには出力ディレクトリを指定します。
msatojson -i /path/to/a3m_containing/directory -o /path/to/output/directory
現在、テンプレート構造情報のインクルードはサポートされていません。
ここで、P9WIC5.jsonが生成されます。このファイルを用いて、次にリガンド情報を追加します。
予測したいリガンド分子の情報をJSONファイルに追加する
リガンド分子の入力は、1つにChemical Component Dictionary (CCD)で規定されているIDを使うものがあります。CCDとはProtein Data Bank (PDB)に登録されている化合物の情報をまとめたデータベースです。こちらは3文字最大5文字で入力する化合物のIDのことで、例えば"ATP"であったりheme-c分子である"HEM"などがあります。CCDに登録されている化合物の情報検索は、Chemical Components in the PDBやRCSB PDBで行うことができます。ここに登録されている化合物があれば、AlphaFold3のinput JSONにCCDを指定することでそのリガンドとの複合体予測をさせることが簡単に行えます。
2024年頃に、従来の3文字表記でのCCD IDは文字パターンを使い切ってしまったため、現在は5文字の化合物IDが発行されるようになりました。例:A1A06 https://www.rcsb.org/ligand/A1A06
なお、AlphaFold3の実行時にCCDとして参照可能なリガンド情報は、各AlphaFold3が利用している仮想環境pythonの中のpython3.xx/site-packages/share/libcifpp/components.cifというファイルにまとめて書かれています。
しかし、当然ですがすべての化合物がCCDとして登録されているわけではありません。その場合はどうすればよいでしょうか?
2つ方法があり、1つはSMILES形式で指定する方法、もう1つはUser-provided CCDを作成してそれを指定させる方法です。特に、タンパク質に対して共有結合性のリガンドを予測させたい場合はSMILESではなく(User-provided) CCDでなければ指定不可であることに気をつけてください。
ここではUniProt ID: P9WIC5のChorismate pyruvate-lyaseの酵素基質複合体を予測するために、リガンドとしてChorismate(コリスミ酸)の情報を追加する方法を紹介します。
1. SMILES形式で指定する
化合物のSMILES形式で入力する方法が最も簡便です。SMILES形式は化合物を文字列で表現するものです。任意の化合物のSMILES形式の表現を見つけたい場合は、ChEBIまたはPubChemなどの化合物データベースを使うと簡単に見つけることができます。または、ChemDrawなどを持っている場合は、その機能の中に存在しています。
ChEBIの検索窓に"Chorismate"と入力すると、SMILES形式はO[C@@H]1C=CC(=C[C@H]1OC(=C)C([O-])=O)C([O-])=Oとなっていることがわかります。
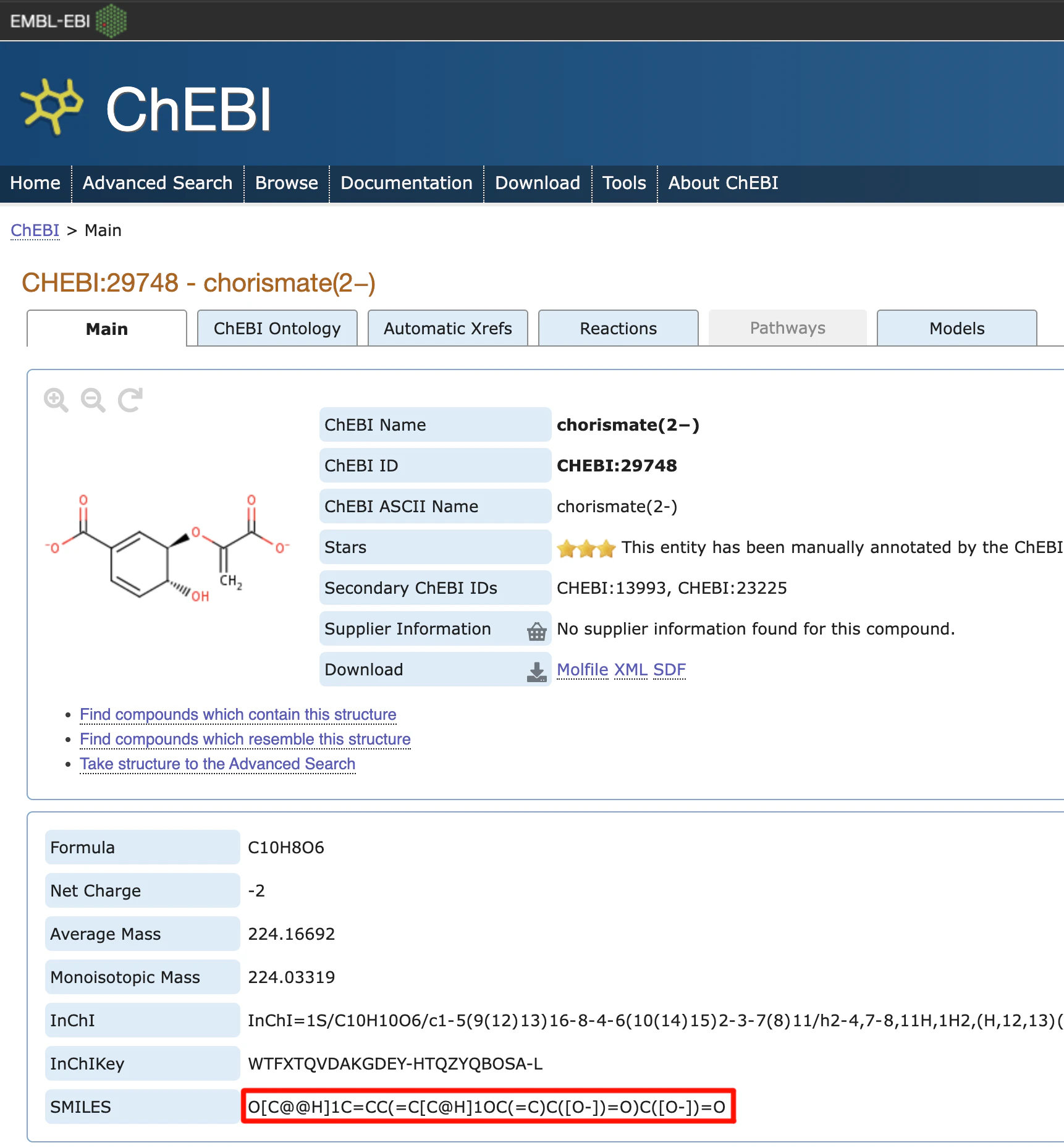
SMILESの文字列がわかったら、modjsonコマンドを使ってJSONファイルにリガンド情報を追加します。
modjson -i P9WIC5.json -o P9WIC5_chorismate_smiles.json -a smiles "O[C@@H]1C=CC(=C[C@H]1OC(=C)C([O-])=O)C([O-])=O" 1 -n P9WIC5_chorismate_smiles
-aには追加するリガンドの情報を指定します。ここではsmilesを指定しています。-aは3つ組の指定を要求しており、1番目はsmilesまたはccdCodesのどちらかの文字列を、2番目はSMILES文字列、またはccdのコード文字列を入れます。3番目にはいくつの分子を導入したいかを整数値で指定します。-a smiles "CCC" 1とすると、SMILES形式の"CCC"を1分子追加することになります。SMILESの文字列はダブルクォーテーションで囲む必要があります。
-iには入力JSONファイル名を指定します。-oには出力JSONファイル名を指定します。-nには、この後のAlphaFold3の構造予測時に出力されてほしいディレクトリ名を指定します。
このコマンドを実行することで、P9WIC5_chorismate_smiles.jsonが生成されます。これを用いてAlphaFold3を実行します。
2. User-provided CCDを作成して予測に用いる
User-provided CCDを作成するうえで満たす項目についてはAlphaFold3 input documentationを読んでいただきたいのですが、理想的な座標が含めることが必須となります。理想座標を作るのはめんどくさいですが、もし化合物の情報がPubChemに存在する場合、理想座標をSDF形式でダウンロードすることが可能です。
先程のUniProtのCatalytic activityの画像で赤矢印で示しているリンクを押すとChEBIに飛びますが、そこでさらにAutomatic Xrefsのタブを開きます。
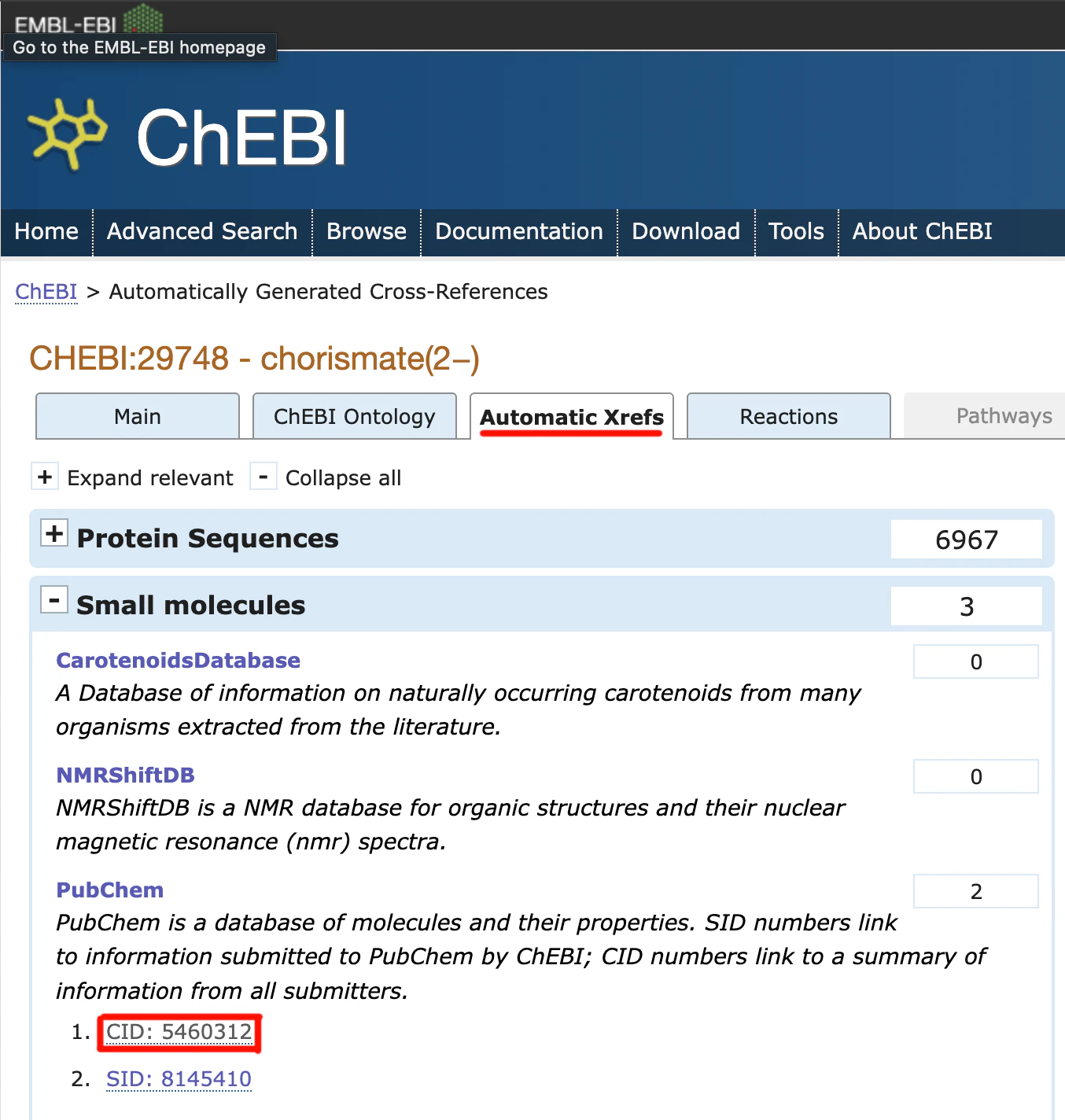
こちらのCIDを押すことでPubChemのページに飛び、3D conformerのDownload CoordinatesからSDF形式のファイルをダウンロードすることができます。
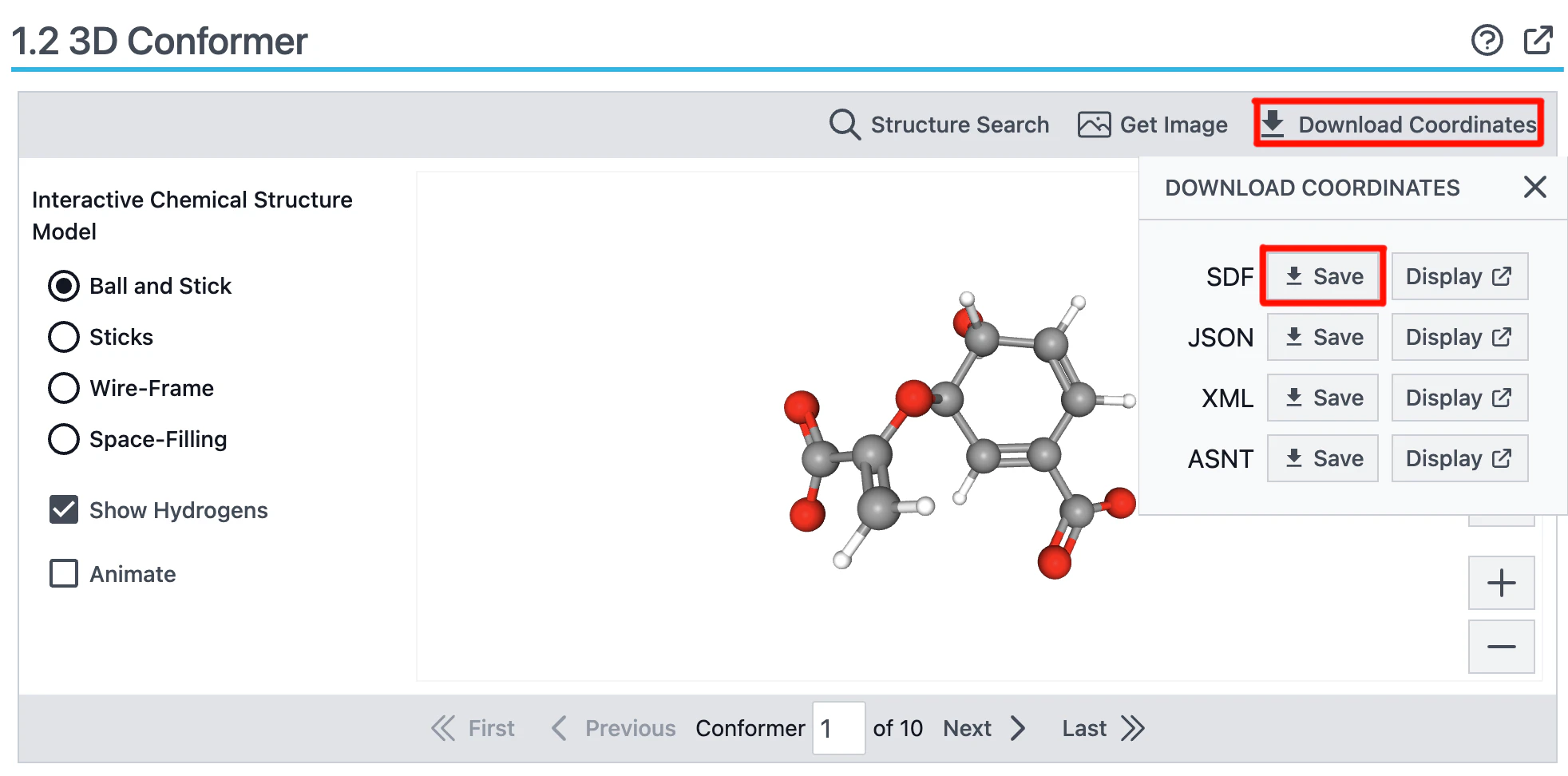
こちらのファイルをダウンロードしたら、sdftoccdコマンドを使ってCCD形式に変換します。
sdftoccd -i Conformer3D_COMPOUND_CID_5460312.sdf -n CHO -o chorismate.ccd
-iにはダウンロードしたSDFファイルを指定します。-nにはCCDの名前を指定します。-oには出力ファイル名を指定します。
-nでのCCDCodeの指定は6文字以内の大文字英字で指定してください(3文字が推奨)。なお、ここではchorismateの頭文字を取ってCHOとしましたが、これはすでにCCDにGlycochenodeoxycholic acidとして登録されています。しかし、User-provided CCDを使用した場合はそちらが優先される仕様となっているため、これを用いたAlphaFold3の予測構造の中ではchorismateとして扱われます。
このccdファイルを用いて、再びmodjsonコマンドを用いてJSONファイルにリガンド情報を追加します。
modjson -i P9WIC5.json -o P9WIC5_chorismate_ccd.json -n P9WIC5_chorismate_ccd -a ccdCodes CHO 1 -u chorismate.ccd
-iは入力JSONファイル名、-oは出力JSONファイル名、-nは出力ディレクトリ名を指定します。-aは追加するリガンドの情報を指定します。ここではccdCodesを指定しています。-aは3つ組の指定を要求しており、1番目はsmilesまたはccdCodesのどちらかの文字列を、2番目はSMILES文字列、またはccdのコード文字列を入れます。3番目にはいくつの分子を導入したいかを整数値で指定します。-a ccdCodes CHO 1とすると、CCD形式の"CHO"を1分子追加することになります。-uには、先程作成したUser-provided CCDファイル名を指定します。
先述の通り、この"CHO"はUser-providedで指定したchorismateのものになります。これを用いてAlphaFold3を実行します。
AlphaFold3で構造予測を実行
こうして出力されたJSONファイルを用いてAlphaFold3を実行します。AlphaFold3のインストールと実行コマンドについては以前の記事を参照してください。
alphafold3実行コマンドのpython3.11 ${ALPHAFOLD3DIR}/run_alphafold.pyに与える引数--json_pathに、先程作成したJSONファイルを指定します。また、--run_data_pipeline=Falseを指定することで、MSA取得処理をスキップし、構造予測処理部分だけを実行させることができます。
# ALPHAFOLD3DIRなどディレクトリ変数名などの定義は省略しています
# Qiita記事の方を参照してください
python3.11 ${ALPHAFOLD3DIR}/run_alphafold.py \
--run_data_pipeline=False \
--jackhmmer_binary_path="${HMMER3_BINDIR}/jackhmmer" \
--nhmmer_binary_path="${HMMER3_BINDIR}/nhmmer" \
--hmmalign_binary_path="${HMMER3_BINDIR}/hmmalign" \
--hmmsearch_binary_path="${HMMER3_BINDIR}/hmmsearch" \
--hmmbuild_binary_path="${HMMER3_BINDIR}/hmmbuild" \
--db_dir="${ALPHAFOLD_DB_DIR}" \
--json_path="P9WIC5_chorismate_ccd.json" \
--model_dir="${MODEL_DIR}" \
--output_dir="${OUTDIR}"
これで構造予測を実行させます。結果はOUTDIRディレクトリに保存され、modjsonのときの-nで指定した名前のサブディレクトリに保存されます。(なお、サブディレクトリ名はAlphaFold3のコードによって自動的に小文字に変換されています)。
p9wic5_chorismate_ccd
├── p9wic5_chorismate_ccd_confidences.json
├── p9wic5_chorismate_ccd_data.json
├── p9wic5_chorismate_ccd_model.cif
├── p9wic5_chorismate_ccd_summary_confidences.json
├── ranking_scores.csv
├── seed-1_sample-0
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
├── seed-1_sample-1
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
├── seed-1_sample-2
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
├── seed-1_sample-3
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
├── seed-1_sample-4
│ ├── confidences.json
│ ├── model.cif
│ └── summary_confidences.json
└── TERMS_OF_USE.md
予測結果の確認
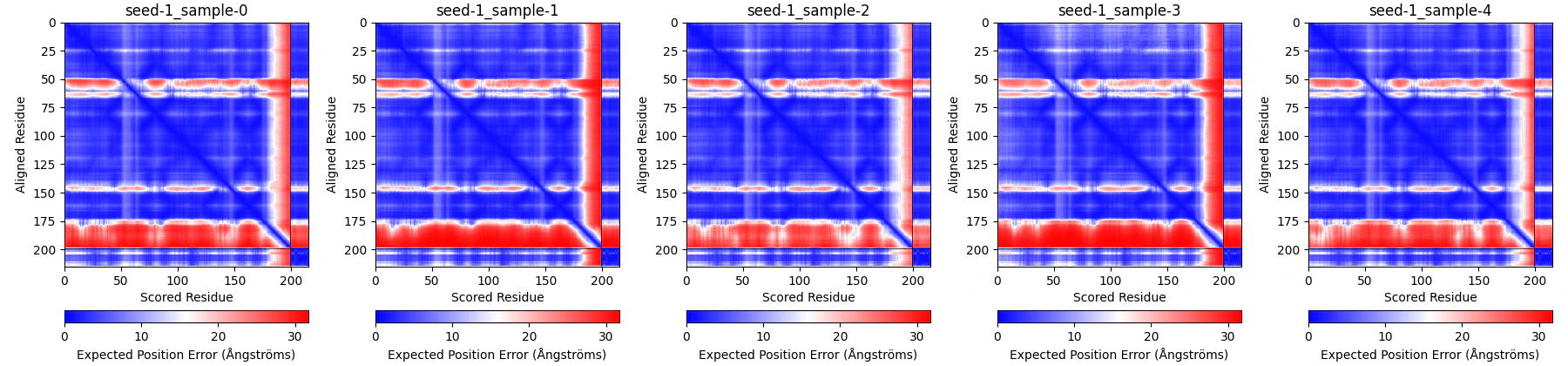
以前の記事にて、pLDDTをPyMOLやChimera上で色分けして表示する方法を紹介しました。alphafold3_toolsはこれに加えて、paeplotというコマンドを使うことで、Predicted Aligned Error (PAE)を画像として出力することもできます。
# /path/to/p9wic5_chorismate_ccd は先程AlphaFold3でアウトプットされたディレクトリへのパス
paeplot -i /path/to/p9wic5_chorismate_ccd [-c Greens_r] [--dpi 200] [-n foo] [-a]
-iには出力ディレクトリ内のサブディレクトリp9wic5_chorismate_ccdへのパスを指定してください。[]の中は任意のオプションです。やっていることは、これの中身の*_confidences.jsonファイルを読み取り、その中のPAEの値を画像にしています。-cでカラーマップを指定できます。デフォルトはbwrです。--dpiで解像度を指定できます。-nで出力ファイル名を指定できます。-aをつけると、すべてのseed-*-sample-*の図を出力します。
PAE画像はインプットディレクトリ/path/to/p9wic5_chorismate_ccdの中にp9wic5_chorismate_ccd_af3pae.pngとして出力されます。
また、すべての出力mmCIFファイルを重ね合わせて1つのmodel.cifファイルに出力するコマンドsuperpose_ciffilesもあります。同様に、-iには出力ディレクトリ内のサブディレクトリp9wic5_chorismate_ccdへのパスを指定してください。
superpose_ciffiles -i /path/to/p9wic5_chorismate
重ね合わせた結果は-iで指定したディレクトリ内に**_superposed.cifというファイル名で出力されます。これをPyMOLで開いてみると、すべてのモデルが重ね合わされていることが確認できます。
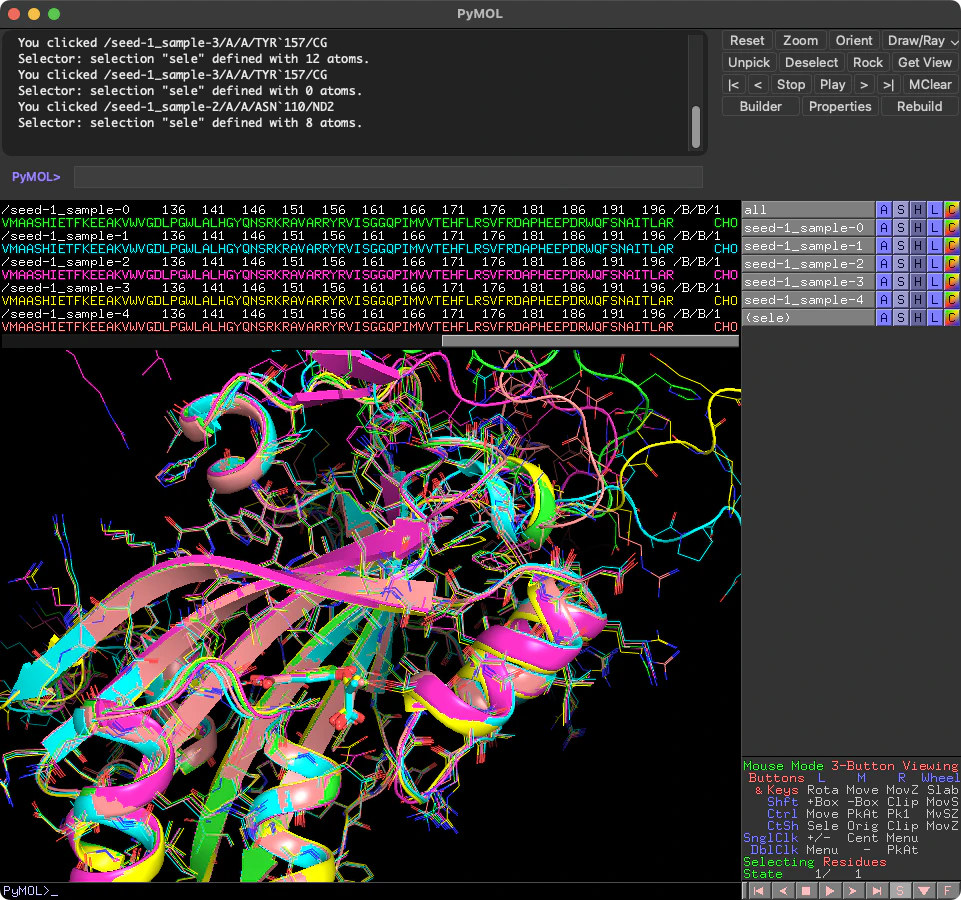
これの何が嬉しいかというと、AlphaFold3はデフォルトで5つのモデルを出すのですが、キラリティを持つ化合物の場合の構造予測は、確率で入力通りになっていないことが多々あります。そのうちベストの構造がp9wic5_chorismate_ccd_model.cifとして提示されるのですが、実際にはそのキラリティが間違っており、各サブディレクトリseed-*-sample-*のモデルのどれかに正しいキラリティを持つものが予測されていることがあります。そんなとき、これを使ってまとめた構造をPyMOLで見ることで、正しいリガンドの構造を持つものだけを選択することができるようになります。