TCGAデータを用いた発現情報リストの作成
TCGAからの原始的なデータ取得の続きです。
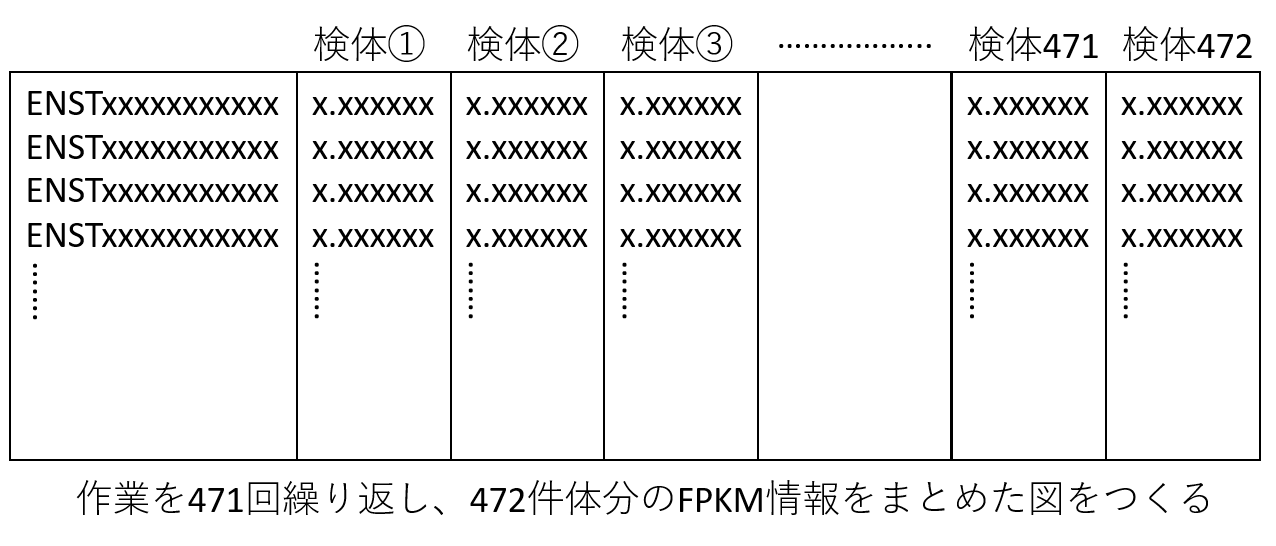
行方向に遺伝子の発現情報、列方向に検体が並ぶ形の表を作ることを目標とします。
今回はTCGAのmelanomaの検体データ472例分なので、60483行、472列の表が最終形態となります。
作りたいもの
FPKMの情報のファイルは、下記のような内容となっている。各行にEnsembl IDとFPKMの値が入っている。ヘッダーはなし。

検体1のファイルの右端に、検体のファイルの2列目(FPKMの情報)をくっつけるという作業をします。
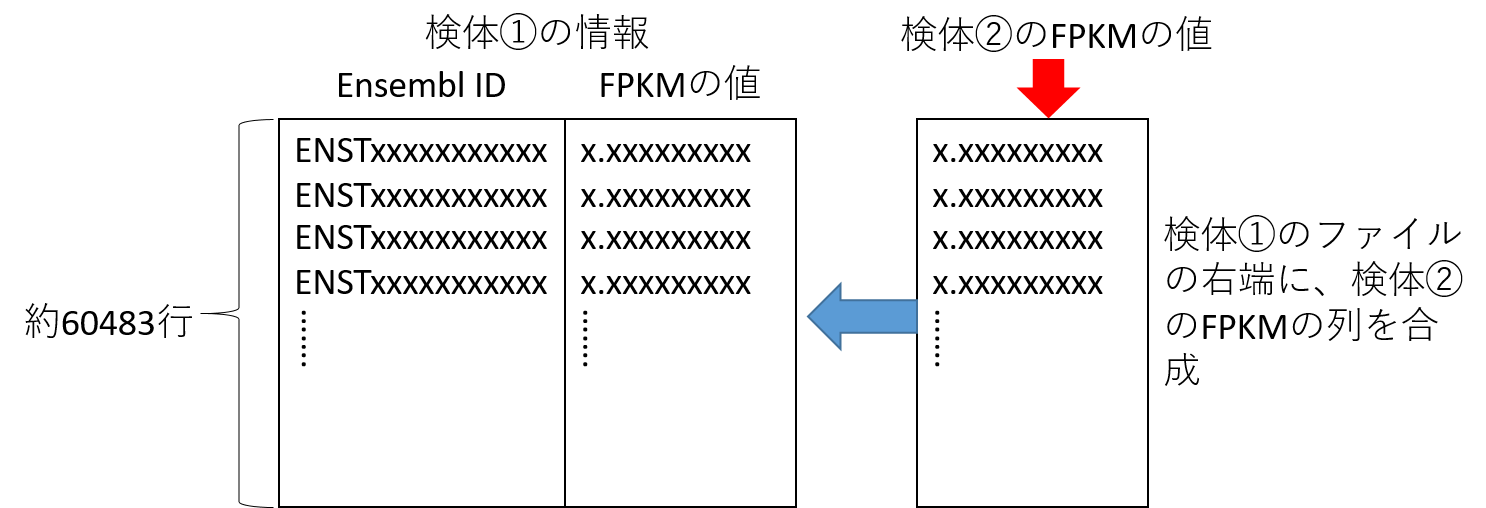
この作業を検体の人数分行い、
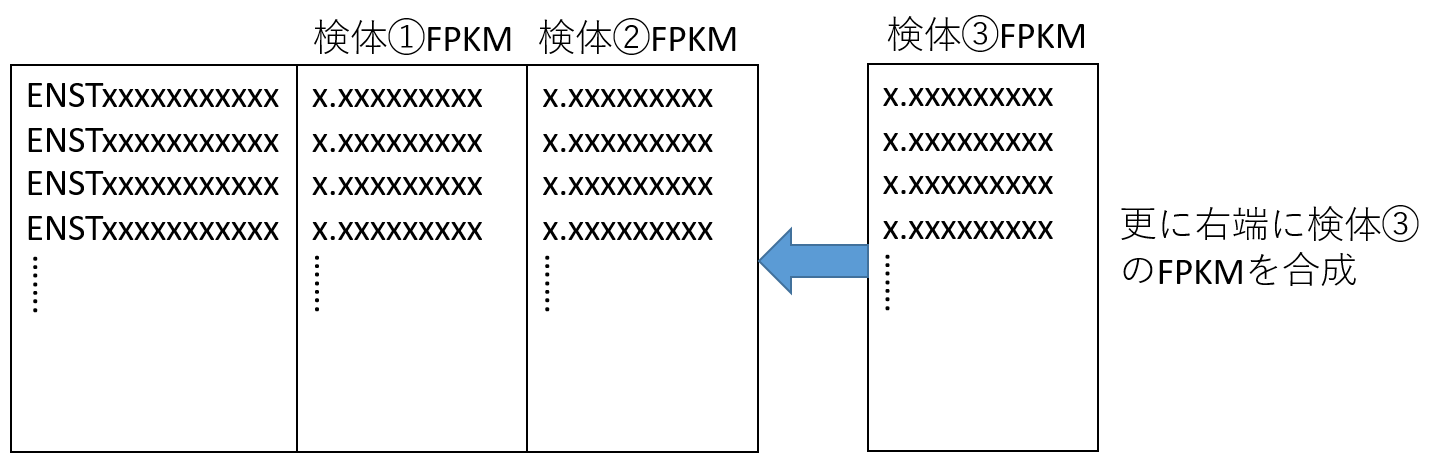
最終的に、全472検体の60483遺伝子の発現情報が含まれるリストを作成する。
現状の確認
現在、作業フォルダの中には、
gdc-client
gdc_manifest.2018-12-03.txt
gdc_sample_sheet.2018-12-17.tsv
の3つと、FPKMの情報が入ったフォルダが472個存在しています。
ファイルの移動と解凍
発現情報のファイルは、各フォルダの中にgz形式で存在しています。そのままでは色々面倒。
なのでひとところにまとめましょう。
mkdir FPKM
mv */*.gz FPKM
# フォルダ内のファイルを全てFPKMフォルダに移動
gunzip FPKM/*.gz
# 解凍
FPKMをまとめたフォルダに移動し、Rを起動。
cd FPKM
sudo R
R version 3.2.3 (2015-12-10) -- "Wooden Christmas-Tree"
Copyright (C) 2015 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu (64-bit)
(中略)
'q()' と入力すれば R を終了します。
>
> list.files()
[1] "00e2c524-8a29-46d7-bf88-ed5f9c3dcf32.FPKM.txt"
[2] "0170ee02-a63a-455c-b41d-bb72f3e515da.FPKM.txt"
[3] "0224b65f-ac08-4f17-b22b-e17bff4d25f4.FPKM.txt"
[4] "02262d0f-84d4-4683-80a4-48c890313132.FPKM.txt"
[5] "0266d0b5-b87f-4356-93b8-c1e291292cc8.FPKM.txt"
...
[471] "ffc004ea-3ab7-41fb-9f49-d1cf8081d982.FPKM.txt"
[472] "fff8635a-798d-400c-83a5-2dd6b6ba5dba.FPKM.txt"
ご丁寧に、ファイル名に「FPKM」と記載してくれているのだが、なぜTCGA IDを付記してくれていないのか理解に苦しむ。
> name <- list.files()
# ファイル名をすべて取り込み、
> exp <- read.table(name[1],header=F)
# ファイル名の1番目に乗っているものをテーブルとして読み込んだ
> dim(exp)
[1] 60483 2
# 60483行、2列のテーブルとして読み込まれていることがわかる。
> head(exp) #先頭を表示させるとこのようになる
V1 V2
1 ENSG00000242268.2 0.000000
2 ENSG00000270112.3 0.000000
3 ENSG00000167578.15 2.262299
4 ENSG00000273842.1 0.000000
5 ENSG00000078237.5 2.689470
6 ENSG00000146083.10 10.189685
FPKMファイルの情報の連結
この「exp」と名付けたテーブルの右側に、他のファイルのFPKM情報をつなげていく。
> n<-length(name);n
[1] 472
> exp<-exp[order(exp[,1]),]
# mergeで表を連結する際には勝手に行がソートされるので、予めEnsemblIDの順に並べ替える
上記の「exp」の右側に、他のFPKMファイルを連結していくスクリプトを作成。
「exp」と同じく、EnsemblIDの順に行を並べ替えて、仮に行数や行の配列が異なっている場合にはその旨を表示させる。
> for (i in 2:n){
exp2<-read.table(name[i],header=F)
exp2<-exp2[order(exp2[,1]),]
ifelse(identical(exp[,1],exp2[,1]),
exp<-merge(exp,exp2,by="V1"),
print("行名がミスマッチ"))
print(ncol(exp))
}
# 「exp」のテーブルの行・列数を確認
> dim(exp)
[1] 60483 473
作業終了後、「50件以上の警告がありました」という趣旨のメッセージが表示される場合があるが、その警告は「列名の重複」を示すもの。列名にはこのあとの行程でTCGA IDを嵌めこむし、表自体はちゃんと作成されるので気に留めなくてもよい。
サンプルシートの読み込み
ファイル名からTCGA IDに変換するため、すでに入手済し、「TCGA-SKCM」フォルダにあるサンプルシートを読み込む。
> sample_sheet<-read.delim("/hoo/bar/TCGA-SKCM/gdc_sample_sheet.2018-12-17.tsv",header=T)
サンプルシートの一部を表示させてみる。2列目の「File.Name」を参考に7列目の「Sample.ID」を前出の「exp」の列名に加えることになる。
> head(sample_sheet)
File.ID
1 38dcc304-2c88-47b7-a5c3-4ec10bc044e9
2 e7967f37-29fb-4192-a888-57afc2df8bff
3 53cce68a-0155-4ab6-aac2-b61a95682695
4 f38a54a1-58b7-4dc5-b0a4-57ce288a93aa
5 7e3679d2-560c-431c-b564-9171aaec09ad
6 a4ad6c5d-02f2-4c7b-813b-93f99b4b5dee
File.Name Data.Category
1 0ef4e36a-8e8a-4d7e-8c4c-e194804d326d.FPKM.txt.gz Transcriptome Profiling
2 92020ebe-72a5-4ccd-8c42-713a0dc51866.FPKM.txt.gz Transcriptome Profiling
3 62532090-07c1-4594-b38e-9d1b1df7a715.FPKM.txt.gz Transcriptome Profiling
4 9d6e0aaf-5b49-4e0f-9fbc-1e9b932ce1c0.FPKM.txt.gz Transcriptome Profiling
5 9943770b-b3e2-42c9-a286-85b558de7da3.FPKM.txt.gz Transcriptome Profiling
6 960ada56-24bd-40ab-91ca-113237e390f9.FPKM.txt.gz Transcriptome Profiling
Data.Type Project.ID Case.ID Sample.ID
1 Gene Expression Quantification TCGA-SKCM TCGA-EB-A6R0 TCGA-EB-A6R0-01A
2 Gene Expression Quantification TCGA-SKCM TCGA-D3-A3CB TCGA-D3-A3CB-06A
3 Gene Expression Quantification TCGA-SKCM TCGA-WE-A8K4 TCGA-WE-A8K4-01A
4 Gene Expression Quantification TCGA-SKCM TCGA-EB-A3XE TCGA-EB-A3XE-01A
5 Gene Expression Quantification TCGA-SKCM TCGA-FS-A1ZQ TCGA-FS-A1ZQ-06A
6 Gene Expression Quantification TCGA-SKCM TCGA-EE-A29G TCGA-EE-A29G-06A
Sample.Type
1 Primary Tumor
2 Metastatic
3 Primary Tumor
4 Primary Tumor
5 Metastatic
6 Metastatic
行名、列名の挿入
このサンプルシートをファイル名の順でソートし、先ほど作った「exp」の列名にサンプルシートの7列目を当てはめるという作業を行う(下図参照)。
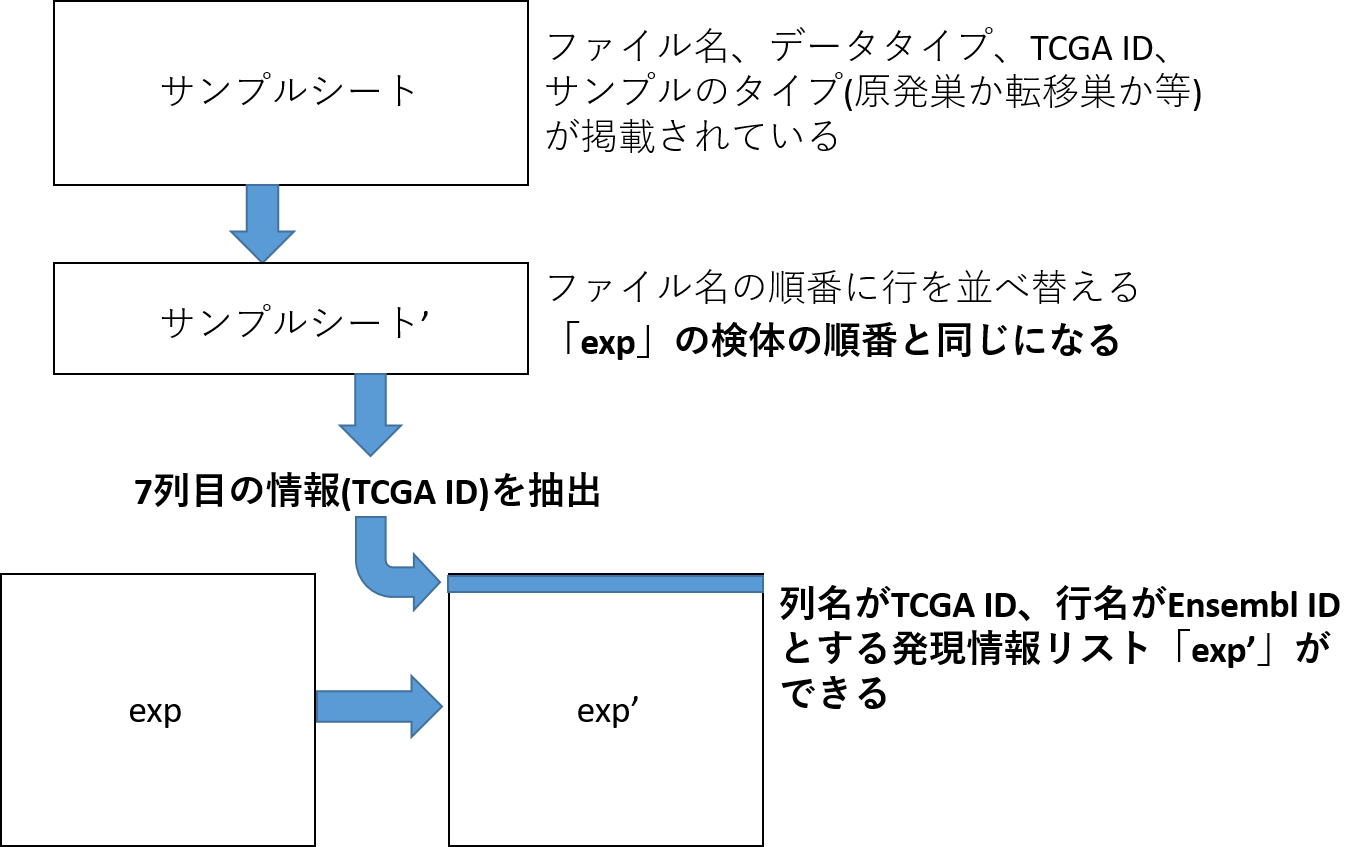
2列目のファイル名のうち、拡張子の「gz」が作業に邪魔なので削っておく。
そのあと、サンプルシートを2列目のファイル名に沿ってソートする。
> sample_sheet[,2]<-gsub(".gz","",as.matrix(sample_sheet[,2]))
> sample_sheet<-sample_sheet[order(sample_sheet[,2]),]
これで、サンプルシートの行の並びは先ほど作った「exp」のファイル順の元となった文字列のグループである「name」の並びと同じになったはずである。
「exp」の作成に使ったファイル名リスト「name」と同一の並びであるかをチェックする。
> identical(as.matrix(name),as.matrix(sample_sheet[,2]))
[1] TRUE
TRUEが出れば作業がうまくいっている。FALSEの場合、並べ変える列を間違えていないか、そもそもファイルが異なっていないかを今一度確認。
「exp」の1列目はEnsembl IDが書かれており、行名は行数となっている。行名にEnsembl IDを入れることをまず行う
> exp[,1] <- substring(exp[,1],1,15)
#Ensembl IDは左から15文字めまでなので、小数点以下を削る
>rownames(exp) <- as.matrix(exp[,1])
#行名に、1列めの情報をあてはめる
>exp<-exp[,-1]
#1列目は不要になったので、削除
続いて、サンプルシートの7列目のTCGA IDを列名に入れる
> colnames(exp) <- as.matrix(sample_sheet[,7])
これで、行名にEnsembl ID、列名にTCGA IDを持つ発現情報リストが出来上がった。
忘れないうちに、保存しておこう。
> write.table(exp,"20181231_TCGA_SKCM_FPKM.txt", sep="\t")
とはいっても、この表は遺伝子名がEnsembl IDでしか表示されていないので、KRASだのTP53だのBRAFだのといったGene symbolでの発現量を得るには更に手間を加える必要がある。
続きは随時追加してまいります。