今回の記事は、20年前に花粉症を抑えるペプチド(薬)をデザインしようとして途中で挫折した経験を振り返る作業
https://note.com/kojikoji3744/n/neddd41a8e645
の続きである。 今回、20年ぶりにin silico screeningのツールに触れた。それは、20年前に使っていた「Tinker+Autodock+RasMol」とは違うツール、「PyMol+HADDOCK」「PyMol+ Rosetta FlexPepDock」である。遅ればせながら、これらのツールについての印象をここに記してみたい
3つのツールの位置づけ比較
- AutoDock / AutoDock Vina
目的:低分子リガンド(薬候補)とタンパク質の結合予測
特徴:
古典的ドッキング(剛体+回転自由度)
受容体は固定 or 部分的に柔軟
スコア関数は経験的(疎水相互作用、H-bond、torsional penalty…)
メリット:軽い・高速、汎用性が高い
デメリット:タンパク質-ペプチドのように「大きく柔軟なリガンド」は苦手
ぼくが 20年前に IgE-Fc と 6残基ペプチドで使ったのはこの系統。最後の時期、これらをGUIで扱えるPyRxが提供され、ぼくのAutoDockを自動化する努力がそこで実現されてしまい少し力が抜けたという記憶がある。
2 HADDOCK (High Ambiguity Driven DOCKing)
目的:タンパク質-タンパク質、タンパク質-ペプチド、タンパク質-DNA/RNA の複合体形成予測
特徴:
情報駆動型(情報があれば拘束条件を活用できる)
柔軟性を持たせた docking(受容体の側鎖やペプチドのバックボーンもある程度動く)
スコア関数はエネルギー(vdW, Elec, Desolvation)+実験的拘束(NMR, MS, Crosslinkなど)が統合可能
メリット:
Web で無料、学術メール不要
ペプチドドッキングに対応
自分の「予想結合部位」を拘束条件に入れると精度UP
デメリット:
完全 ab initio 探索には弱い(入力配置や拘束に依存)
Rosetta ほど詳細なサイドチェーン・原子レベル最適化はしない
AutoDock よりペプチドに強いが、FlexPepDockほど原子レベルの精密性はない
3 Rosetta FlexPepDock
目的:タンパク質-ペプチド相互作用の 高分解能予測
特徴:
受容体:部分柔軟(側鎖、軽いバックボーン)
ペプチド:フル柔軟(全バックボーン+側鎖)
サンプリング:Monte Carlo + fragment insertion
Rosetta スコア関数(物理+統計ポテンシャルの融合)
メリット:
世界的に最も広く使われている「ペプチドドッキング専用」の精密ツール
高分解能リファインメントで、原子レベルの界面構造が得られる
デメリット:
アカデミックライセンス必須(ROSIE も同様)
計算コストが高い(数百〜数千構造を生成 → クラスタリング必須)
ペプチド専用・原子分解能モデル = FlexPepDock の強み。
ということで、当初、Rosetta FlexPepDockを扱う予定でいたが、現時点ではそれを無料でつかうためには「学術メール」が必要と判明したため、それは断念。「学術メール」がないぼくでも無料で扱える HADDOCK をここではみていく。
*もし、ぼくに「学術メール」を提供してくださる方がおられれば、是非連絡ください。お待ちしてます。
[HADDOCK におけるペプチドの扱い]
基本
HADDOCK は タンパク質・ペプチド・DNA/RNA をすべて「ポリマー」扱いする。つまり ペプチドも普通のPDB形式でOK。特別に「小分子化」する必要はない(AutoDock では「低分子扱い」なので、ペプチドを Tinkerなどで事前に加工してリガンド構造に変換 するが、その必要はない)。
具体的には最終的に必要なファイルが次のふたつ:
① リガンド(YFGHWHペプチド) →YFGHWH.pdb を作成
② 受容体(IgE-Fc部分) → 余分な鎖や水、糖鎖を削除したPDB
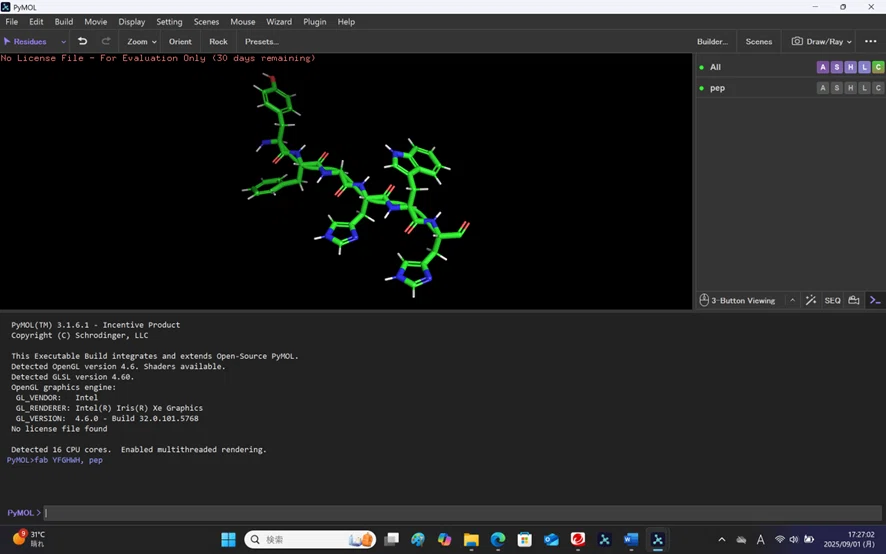
①手順:PyMOLで6残基ペプチドを作る
- PyMOL を起動 評価版でもOK。PyMOLのコマンド入力欄(右下 PyMOL>)を使う。
- ペプチドの生成コマンド
PyMOL には fab(fragment add builder)という便利なコマンドがあります。これを使うと、配列から直鎖ペプチドを簡単に作成できます。
fab YFGHWH, pep
これで「鎖A」の6残基ペプチドが生成されます。 - 鎖IDを変更(HADDOCKでわかりやすくする)
alter pep, chain='P'
HADDOCK に渡すとき「受容体=鎖B,C」「リガンド=鎖P」と明確になるので便利。 - PDBファイルとして保存
save YFGHWH.pdb, pep
(あるいは、絶対パス:
save C:/Users/koji3/デスクトップ/YFGHWH.pdb, pep )
②受容体(IgE-Fc部分) → 余分な鎖や水、糖鎖を削除したPDBをPyMolでつくる
- 1F6Aを取得(ネットが不安定なら "load C:/path/1f6a.pdb" に置換)
fetch 1f6a, async=0
hide everything - IgE-Fc部分だけを作成:鎖B+C(polymer.protein = たんぱく質原子のみ)create ige_fc, (chain B+C and polymer.protein)
- 余分を全削除(水や糖を含むHETATMも全部)
remove not ige_fc
remove hetatm
remove resn HOH - 仕上げ(表示は任意)
show cartoon, ige_fc
color green, ige_fc and chain B
color cyan, ige_fc and chain C - 保存(デスクトップに出したい場合はパスを自分のユーザー名に)
save C:/Users/<あなたのユーザー名>/Desktop/IgE_Fc_clean.pdb, ige_fc
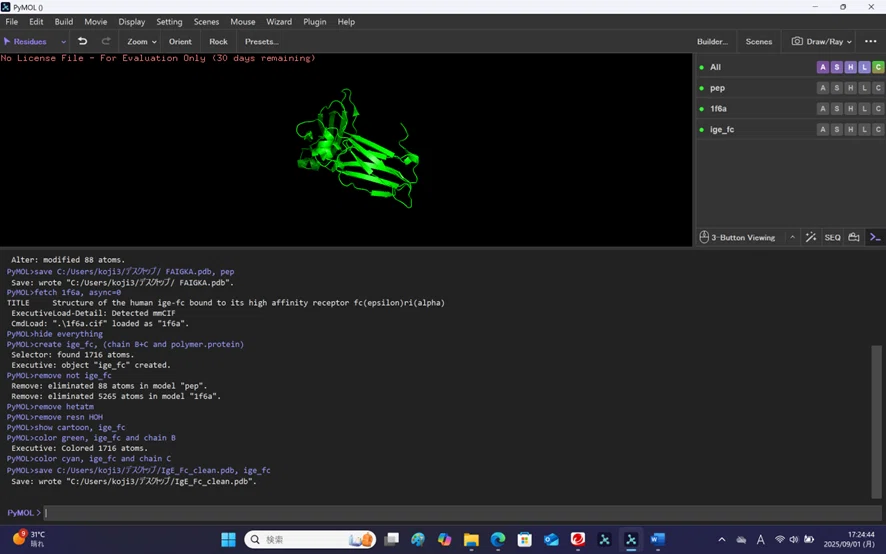
うまくできたかチェック:デスクトップに IgE_Fc_clean.pdb ができる
これで 受容体(IgE-Fc) と ペプチド(YFGHWH.pdb) の2ファイルが揃いました。
次は HADDOCK 2.4 にアップロードして、Active/Passive 残基を指定して投入。
(参考)PyMOL の画面下にある PyMOL> プロンプト(1行の入力欄) が、コマンドを入力できる唯一の場所です。
これは 1行ごと の入力が基本で、長いスクリプトを何行も一度に貼り付けることはできません。長いコマンドをまとめて走らせたいときは .pml スクリプトを用意して @script.pml で読み込む。 あるいは、スクリプトを [File] → [Run Script…] で読み込む
[HADDOCK 2.4 Web 入力手順(IgE-Fc × YFGHWH)]
- サーバにアクセス
HADDOCK 2.4 Webserver
「Submit new job」をクリック - 分子ファイルのアップロード
Molecule 1 (Receptor) → IgE_Fc_clean.pdb
Molecule 2 (Ligand) → YFGHWH.pdb - アクティブ/パッシブ残基の指定
「どの部分が相互作用するか」をある程度ヒントとして与えます。
(1) IgE-Fc 側(Receptor)IgE-Fc の Ce3 ドメイン(FcεRI α鎖が結合する界面)が既知の相互作用パッチです。PDB 1F6A では 鎖 B/C の 330–340 付近が接触部位として有名。
Active 残基例(受容体側)B鎖: 330–340 C鎖: 330–340
だが鎖B/Cを分けて書けないので、番号だけ入力します:
330,331,332,333,334,335,336,337,338,339
(2) ペプチド側(Ligand)YFGHWH は短いので、全残基を「アクティブ」にして問題ありません。
Active 残基例(リガンド側)P:1,2,3,4,5,6
だが、鎖IDは不要で、残基番号だけ:1,2,3,4,5,6 - Passive 残基
HADDOCK の規則では「Active 残基の隣は自動で Passive に補完」されます。なので手動入力は不要。 - その他の設定は初期値
- ジョブ投入
Job name をわかりやすく(例:IgE_YFGHWH_test1)
Submit
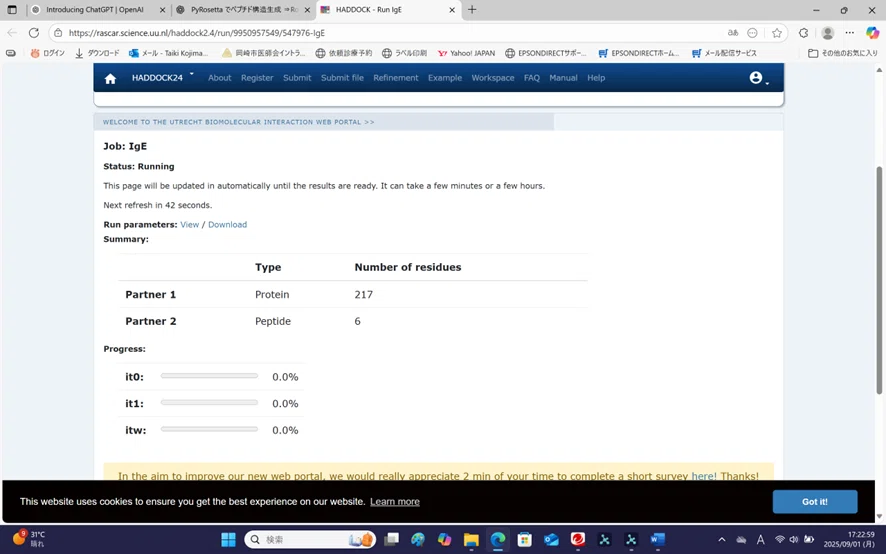
7 出力確認
数時間後、結果ページに(結果へのリンクがメールで届きます)。
Cluster 2 :HADDOCK score-83.2 +/- 4.8 Cluster size37 RMSD from the overall lowest-energy structure0.5 +/- 0.3 Van der Waals energy-39.5 +/- 2.5 Electrostatic energy-118.4 +/- 25.6 Desolvation energy-20.1 +/- 4.6 Restraints violation energy2.0 +/- 1.4 Buried Surface Area868.0 +/- 36.4 Z-Score-2.1
指標の見方: HADDOCK score:低いほど良い Z-score:平均からの標準偏差(より負が良い) Cluster size:大きいほど安定(再現性) 参考値:BSA > ~600 Ų、Restraints violation 小さいのが◎ 今回 Cluster 2:Score -83.2 ±4.8, Z = -2.1, Size 37, BSA 868 Ų, Violation 2.0 → ベスト Cluster 1 は Size 60 と大きいが Score/Z が劣る(-67, Z -0.8)
[In silico 導入についての注意]
どこまで信頼?
例えば、Alpha Foldではタンパク質の立体構造の予想をおこなったが、これには「結晶化で確認された立体構造」という答えがあった。正解があるため、「学習させる」こともできれば、「学習させないにしても自己学習の目標の設定」が容易にできた。
だが、分子間の結合の「答え」はin vitroの結合試験でいいのだろうか?例えば、疎水性の強いペプチドは、水溶液の中では、その結合力はin vitroで正確には測れない。
また、分子間の結合様式には、低分子化合物の「鍵と鍵穴」のようなもの、そして抗体医薬品のような「分子表面での結合」、の大きくふたつがある。
今のin silicoによる、薬の設計は①のみに特化している。というより、最初から②の開発はその視野にない。また、②の結合様式を見据えたペプチド型の薬剤は、「特異性が弱く」「結合性が弱い」ことで最初から視野にない。それは正しい。だが、正しいがゆえに「見なおすべき、暗黙の前提」なのではないか?
たとえば、ペプチド薬剤は、静脈投与でも、経口投与でも、すぐ分解されているので、皮下注(あるいは今回の目標のような、鼻腔内投与)でなければならない、との固定観念は日本ではゆるぐことはない。
ところが、外国からは、たとえば糖尿病治療薬リベルサスという、胃で吸収されるペプチド型の薬剤が開発され、日本でもあっさり認可されたという事実
https://passmed.co.jp/di/archives/579
を基礎の研究者も知り、ペプチドに関する多くの固定観念を見直していくべきではないだろうか?
いずれにせよ、(Autodockの開発速度は鈍ったが)、HADDOCK、Rosetta FlexPepDockは現在も開発が進んでいる一方、まだ、十分Alpha Fold出現によるインパクトが出尽くしていない印象である。残念ながら「ブラックボックス」を知る力のないばくは、新たなソフトをつくることはできず、できあがってきたソフトをあやつることしかできないが、今後もこれらの行方を注視していこうと思う。