背景
諸事情で古いEnsemblのGTFを使っています.
CufflinksとHTSeqの出力を比較した際,Cufflinksでは出力されない遺伝子IDがいくつか見つかりました.それらの遺伝子IDをみてみますと,別の遺伝子IDにアサインされているtranscriptが全く同一の遺伝子構造を持っていることがわかりました.
つまり,以下のことが分かりました(常識?).
- Ensemblの(少なくとも)古いバージョンの遺伝子アノテーションには,本来マージされるべきtranscriptが別の物として登録されている
- そのような'重複した'transcriptのエントリをCufflinksとHTSeqでは異なる取り扱いをする
-- Cufflinksでは,非明示的にそれらをマージし,片方のIDだけ出力する
-- HTSeqでは,別々のtranscriptとして扱う
そこで,Rでそのようなtranscriptがどのくらいあるかを調べてみました.
準備
source("http://bioconductor.org/biocLite.R")
biocLite("TxDb.Mmusculus.UCSC.mm10.ensGene")
# 遺伝子アノテーションのロード
library("TxDb.Mmusculus.UCSC.mm10.ensGene")
# オブジェクト名の確認
ls("package:TxDb.Mmusculus.UCSC.mm10.ensGene")
ensGene <- TxDb.Mmusculus.UCSC.mm10.ensGene
findOverlapがcircular seqではエラーがでるので,ミトコンドリア配列をオフにします.
isActiveSeq(ensGene) <- c(chrM = F)
挙動確認用.
length(transcripts(ensGene)) == length(exonsBy(ensGene, by = "tx"))
## [1] TRUE
transcriptの座標から候補を絞り込む
まず,transcriptのstartとendが同一のtranscriptがあるものを探します.ここではエキソン構造が異なる場合も含まれます.
tx <- transcripts(ensGene)
dup <- countOverlaps(tx, tx, type = "equal") > 1
tx.dup <- tx[dup]
hit <- findOverlaps(tx.dup, tx.dup, type = "equal")
hit <- hit[queryHits(hit) != subjectHits(hit), ]
table(dup)
## dup
## FALSE TRUE
## 89589 5907
head(tx.dup$tx_name)
## [1] "ENSMUST00000061280" "ENSMUST00000172291" "ENSMUST00000166384"
## [4] "ENSMUST00000168907" "ENSMUST00000119714" "ENSMUST00000163561"
hit
## Hits of length 9246
## queryLength: 5907
## subjectLength: 5907
## queryHits subjectHits
## <integer> <integer>
## 1 1 2
## 2 2 1
## 3 3 4
## 4 4 3
## 5 5 6
## 6 6 5
## 7 7 8
## 8 8 7
## 9 9 10
## ... ... ...
## 9238 5899 5898
## 9239 5900 5901
## 9240 5901 5900
## 9241 5902 5903
## 9242 5903 5902
## 9243 5904 5905
## 9244 5905 5904
## 9245 5906 5907
## 9246 5907 5906
エキソン構造を比較する
次にエキソン構造が一致しているものを取り出します.
ex <- exonsBy(ensGene, by = "tx")
dup.ex <- countOverlaps(ex, ex, type = "within", maxgap = 0) > 1
table(dup.ex)
## dup.ex
## FALSE TRUE
## 79223 16273
# ex からtranscript_idでGRangesを取り出せるように
names(ex) = tx$tx_name
# ちょっと高速化
library(multicore)
iden <- mclapply(1:length(hit), function(x) {
a <- queryHits(hit)[x]
b <- subjectHits(hit)[x]
# startとendのデータフレームにした上で,同一かどうか比較
identical(as.data.frame(ex[[tx.dup$tx_name[a]]])[, 2:3], as.data.frame(ex[[tx.dup$tx_name[b]]])[, 2:3])
})
table(unlist(iden))
##
## FALSE TRUE
## 9202 44
結果
hit.identical <- hit[unlist(iden), ]
hit.identical <- hit.identical[queryHits(hit.identical) < subjectHits(hit.identical),
]
pair <- data.frame(a = tx.dup$tx_name[queryHits(hit.identical)], b = tx.dup$tx_name[subjectHits(hit.identical)])
head(pair)
## a b
## 1 ENSMUST00000055830 ENSMUST00000159201
## 2 ENSMUST00000126634 ENSMUST00000134468
## 3 ENSMUST00000056136 ENSMUST00000163374
## 4 ENSMUST00000036819 ENSMUST00000163908
## 5 ENSMUST00000097080 ENSMUST00000114295
## 6 ENSMUST00000051845 ENSMUST00000168574
試しに一つのペアを表示してググってみます.
pair[3, ]
## a b
## 3 ENSMUST00000056136 ENSMUST00000163374
ex[[as.character(pair[3, 1])]]
## GRanges with 2 ranges and 3 metadata columns:
## seqnames ranges strand | exon_id exon_name
## <Rle> <IRanges> <Rle> | <integer> <character>
## [1] chr1 [172341210, 172341451] + | 8917 <NA>
## [2] chr1 [172368921, 172374085] + | 8920 <NA>
## exon_rank
## <integer>
## [1] 1
## [2] 2
## ---
## seqlengths:
## chr1 chr2 ... chrUn_JH584304
## 195471971 182113224 ... 114452
ex[[as.character(pair[3, 2])]]
## GRanges with 2 ranges and 3 metadata columns:
## seqnames ranges strand | exon_id exon_name
## <Rle> <IRanges> <Rle> | <integer> <character>
## [1] chr1 [172341210, 172341451] + | 8917 <NA>
## [2] chr1 [172368921, 172374085] + | 8920 <NA>
## exon_rank
## <integer>
## [1] 1
## [2] 2
## ---
## seqlengths:
## chr1 chr2 ... chrUn_JH584304
## 195471971 182113224 ... 114452
結果の検証
調べてみると,最新のEnsemblアノテーションではマージされているようです.
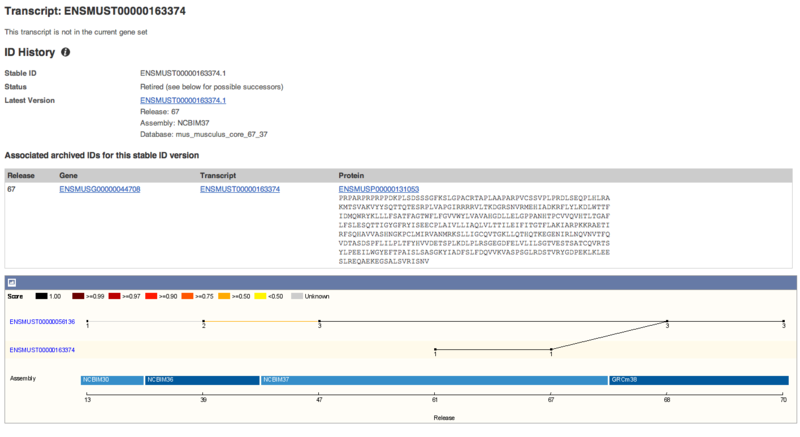
Bioconductorのアノテーションパッケージは最新のものではないのでしょう.
では,最新のアノテーションではこのようなことはないのでしょうか.
調べてみましょう.
EnsemblからTranscripDbオブジェクトを作成
mmusculusEnsembl <- makeTranscriptDbFromBiomart(biomart = "ensembl", dataset = "mmusculus_gene_ensembl")
isActiveSeq(mmusculusEnsembl) <- c(MT = F)
tx.new <- transcripts(mmusculusEnsembl)
dup.new <- countOverlaps(tx.new, tx.new, type = "equal") > 1
tx.new.dup <- tx.new[dup.new]
hit <- findOverlaps(tx.new.dup, tx.new.dup, type = "equal")
hit <- hit[queryHits(hit) != subjectHits(hit), ]
table(dup.new)
## dup.new
## FALSE TRUE
## 87109 3810
head(tx.new.dup$tx_name)
## [1] "ENSMUST00000166384" "ENSMUST00000168907" "ENSMUST00000142117"
## [4] "ENSMUST00000179089" "ENSMUST00000177608" "ENSMUST00000180062"
ex.new <- exonsBy(mmusculusEnsembl, by = "tx")
dup.ex.new <- countOverlaps(ex.new, ex.new, type = "within", maxgap = 0) > 1
table(dup.ex.new)
## dup.ex.new
## FALSE TRUE
## 77370 13549
names(ex.new) = tx.new$tx_name
library(multicore)
iden.new <- mclapply(1:length(hit), function(x) {
a <- queryHits(hit)[x]
b <- subjectHits(hit)[x]
identical(as.data.frame(ex.new[[tx.new.dup$tx_name[a]]])[, 2:3], as.data.frame(ex.new[[tx.new.dup$tx_name[b]]])[,
2:3])
})
table(unlist(iden.new))
##
## FALSE TRUE
## 6264 18
結果
hit.identical.new <- hit[unlist(iden.new), ]
hit.identical.new <- hit.identical.new[queryHits(hit.identical.new) < subjectHits(hit.identical.new),
]
pair.new <- data.frame(a = tx.new.dup$tx_name[queryHits(hit.identical.new)],
b = tx.new.dup$tx_name[subjectHits(hit.identical.new)])
head(pair.new)
## a b
## 1 ENSMUST00000142117 ENSMUST00000179089
## 2 ENSMUST00000055830 ENSMUST00000159201
## 3 ENSMUST00000126634 ENSMUST00000134468
## 4 ENSMUST00000171570 ENSMUST00000179386
## 5 ENSMUST00000066859 ENSMUST00000111976
## 6 ENSMUST00000110186 ENSMUST00000110188
結果の検証
ありました… Ensemblのウェブサイトでも確認できました.
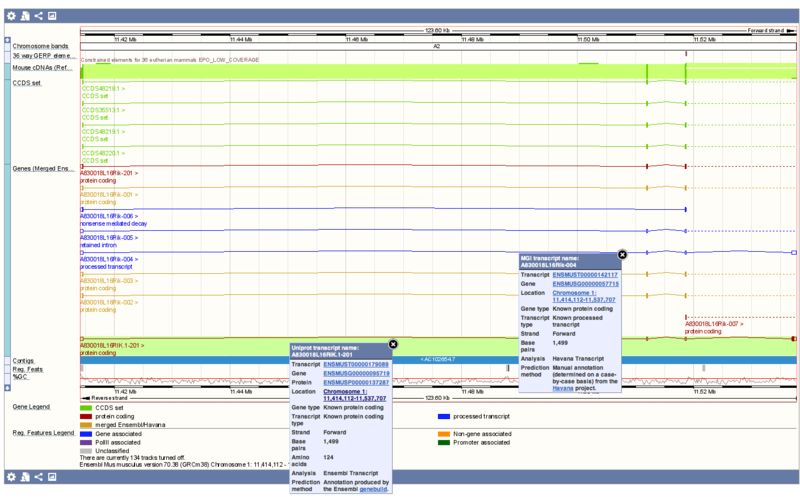
とりあえず,皆さまEnsemblアノテーションを使う場合にはご留意を.