32 99 chrA 1758 60 100M = 1995 337 CGACCTATTACCCAGCCATAAGTTGACAACCAAATCCCTTAAACGCACTGTCTATAAGCGCATATAGTAGACGGACCGTTATCCTCATTAAGGACCATTA 5555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555 NM:i:2 MD:Z:67A21C10
18 147 chrA 1762 60 65M3D35M = 1519 -343 CTATTACCCAGCCATAAGTTGACAACCAAATCCCTTAAACGCACTGTCTATAAGCGCATATAGAAGGACCGTTATCCTCATTCAGGACCATTAATACTGC 5555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555 NM:i:3 MD:Z:65^GAC35
samファイルにはmapping後のreadが保持されている spec
参考1
一行に1つのリードが書いてある。
それぞれのリードがリファレンスと比べて、どんな差を持っているか知りたい。
例えば、1番上のリードは2つ変異がある。
1824-1825, A=>T
1846-1847, C=>A
各列は、順に、seq name,flag(2 bitで表現されている),chr,pos,map quality,CIGAR,seq name of mate read,pos of mate read, insert size, sequence, basequality, tags... となっているspec参照。
CIGAR
CIGARはinsertion, deletion, softclipなどの位置を表す。
例えば、indelがない場合100M (リード長=100bpの場合),途中にdelがある場合30M3D70M、insの場合25M5I70Mのよう。
更に、
N : skipped bases on reference
S : soft clipping
H : hard clipping
P : padding
も使われる。Soft clipはsequenceの項に含まれているがalignしなかったもの、hard clipはalignせずsequenceの項にも含まれていない部分を指す。soft clipped readsはIGVで見るとミスマッチが連なって見える。(img)https://www.google.co.jp/search?q=soft+clipping+reads&client=safari&rls=en&source=lnms&tbm=isch&sa=X&ei=ml_0UqKrCofYkAWL9oGoDg&ved=0CAkQ_AUoAQ&biw=1367&bih=803#q=soft+clipped+reads&rls=en&tbm=isch
注意点は、posはposition of left-most aligned baseなので、soft clipされた部分は含まない。(もちろんhard clipも含まない)
なので、
TCGATCGA
1 2
9Mの場合はposは上の1を指す、3S6Mの場合は2の部分を指す。
MD tag
MD tag は 61G22C15や65^GAC35のようなタグで、リファレンス配列を見ずに、ミスマッチやDeletionの配列を確認できる。(参考)[http://onetipperday.blogspot.jp/2012/07/deeply-understanding-sam-tags.html]
61G22C15であれば、61ベースリファレンスと一致(もしくはInsertion)の後、リファレンスではGだがreadでは"何か"に変わっている。その後、22ベース一致(もしくはInsertion)し、リファレンスがCのミスマッチがあり、その後15ベース一致(もしくはInsertion)している。
65^GAC35の^GACはリファレンス配列のGACの部分がdeletionになっている事を示す。
Insertionの情報はMD tagには含まれない (抜かれている)。なので、リードの各ベースのリファレンス配列における位置を確認する場合は、CIGARと組み合わせる必要がある。
ちなみに、もしMD tagが抜けている場合は、samtools calmdで補完してくれる。
1-basedと0-based
リファレンス配列に対する、ゲノム上の位置を示す場合に、1-basedと0-based positionがある。
引用:
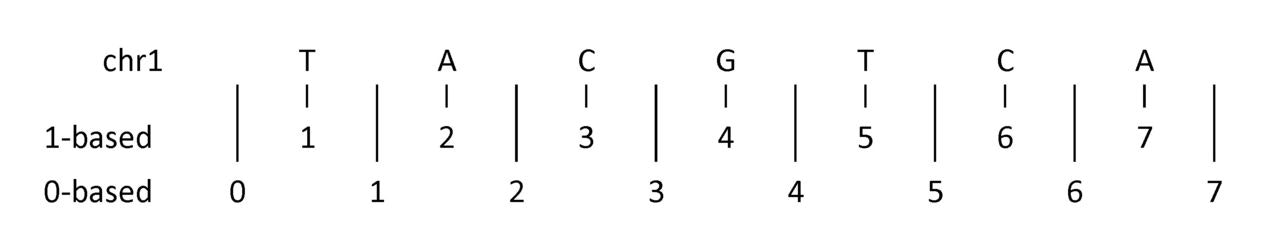
大変わかり易い資料
sam fileは1-based positionを使っている。
今回は変異を表すのに、0-based positionを使う。
MDとCIGARを組み合わせて変異を抽出
少しややこしくて、こんな感じのコードになった。
https://github.com/usuyama/variant_parser/blob/master/sam_parser.rb
あんまり自信ない。
メインの部分は下のような感じ。
@variants = []
while cigar_idx < cigar.length and md_idx < md.length
match([current_cigar.type, current_md.type]) do
with _[:soft, _] {
@variants << current_cigar.clone
get_next_cigar.call
}
with _[_, :del] {
current_cigar.ref = current_md.ref
@variants << current_cigar.clone
get_next_cigar.call
get_next_md.call
}
with _[_, :mismatch] {
@variants << Variant.new({:type => :mismatch,
:ref => current_md.ref,
:len => 1})
current_cigar.len -= 1
get_next_md.call
}
with _[:ins, :match] {
@variants << current_cigar.clone
get_next_cigar.call
}
with _[:match, :match] {
if current_cigar.len > current_md.len
@variants << Variant.new({:type => :match, :len => current_md.len })
current_cigar.len -= current_md.len
get_next_md.call
get_next_cigar.call if current_cigar.len == 0
else
@variants << current_cigar.clone
current_md.len -= current_cigar.len
get_next_cigar.call
get_next_md.call if current_md.len == 0
end
}
end
end
C++ でも実装しました。 こちら