はじめに
ここでは Bayes Linuxには Galaxy がインストールされています。Galaxy上には、RNA-seq解析ワークフローが、組み込まれています。ここではデモデータを利用してRNA-seq解析を実施する方法を述べます。
ワークフローの概要
RNA-seqで得られた配列から遺伝子発現を定量します。発現量の定量には2つの方法があります。ひとつはゲノムに配列をマッピングする方法、もうひとつはトランスクリプトームへマッピングする方法です。Galaxyには後者の方法を採用したワークフローが登録されています。デモデータはマウスES細胞の1細胞RNA-seqのデータになります。
デモデータの入手
まず、以下のいずれかを読み、Bayes Linuxを起動させます。
次にデモデータをインストールします。デモデータは理研上のサーバから入手されます。環境によって数時間程度かかります。
Bayes Linuxにログインするか、VirtualBox上から、以下のコマンドを入力します。
curl -ssL https://bioinformatics.riken.jp/bayes/vm/dl.sh | bash -s
このコマンドにより /data にデータが保存されます。
保存されるデータは以下の通りです。
- adapter_primer (混入が予想されるPCRプライマー配列)
- bowtie2_index (Ensembl transcriptのbowtie2インデックスファイル)
- quartz_div100_rename (RNA-seqデータ, fastq)
- sailfish_index (Ensembl transcriptのsailfishインデックスファイル)
- transcriptome_ref_fasta (Ensembl transcript, FASTA)
Galaxyへのデータの配置
デモデータをGalaxyのデータライブラリへの登録します。
-
Galaxyに、
ID: admin@galaxy.org
パスワード: admin
でログインする -
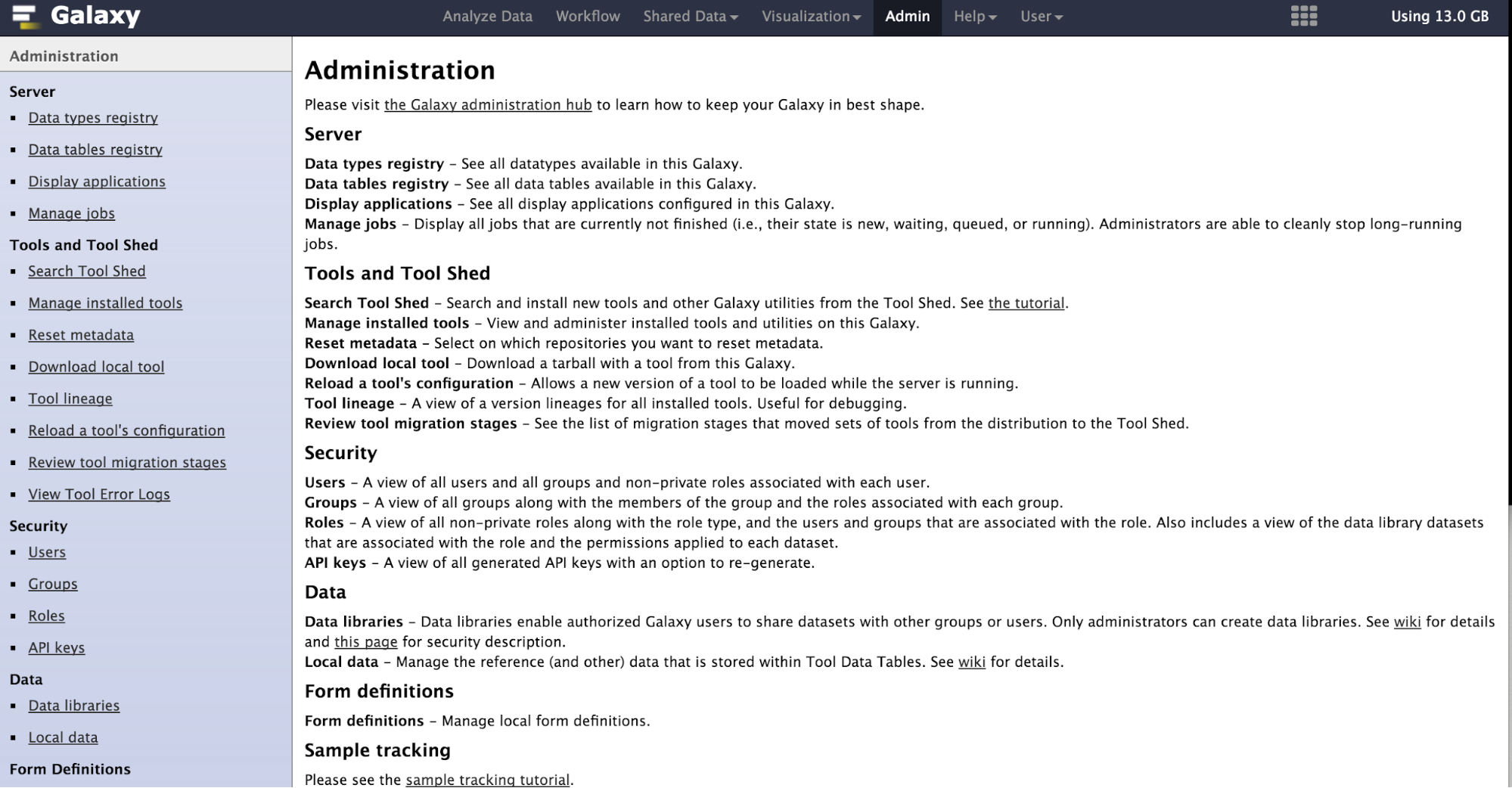
上部のAdminをクリックする

-
左メニューの中の、Data Libraryをクリックする
-
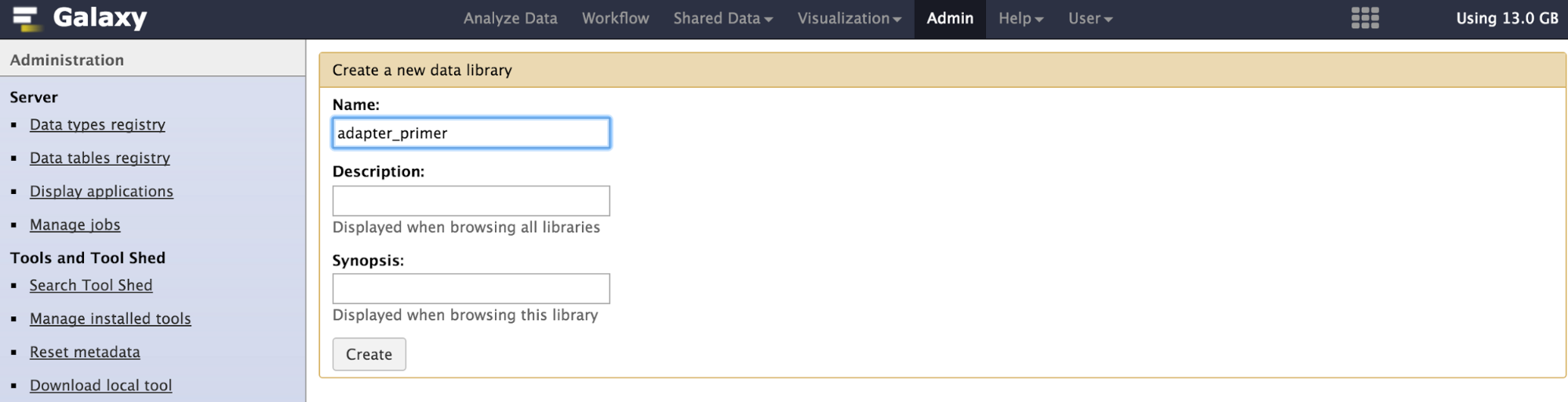
右上の、Create new data libraryをクリックする
-
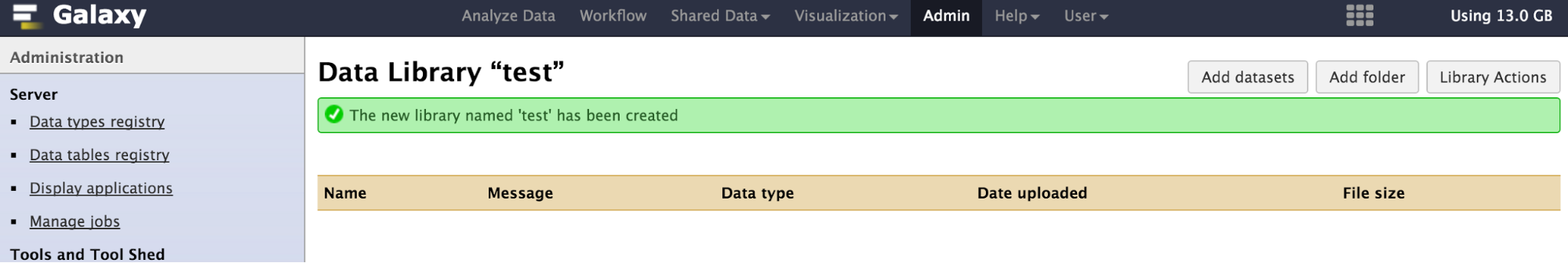
Nameにadapter_primerと入力し、Createをクリックする
-
右上Add datasetsをクリックする
-
以下の情報を入力し、Upload to libraryをクリックする
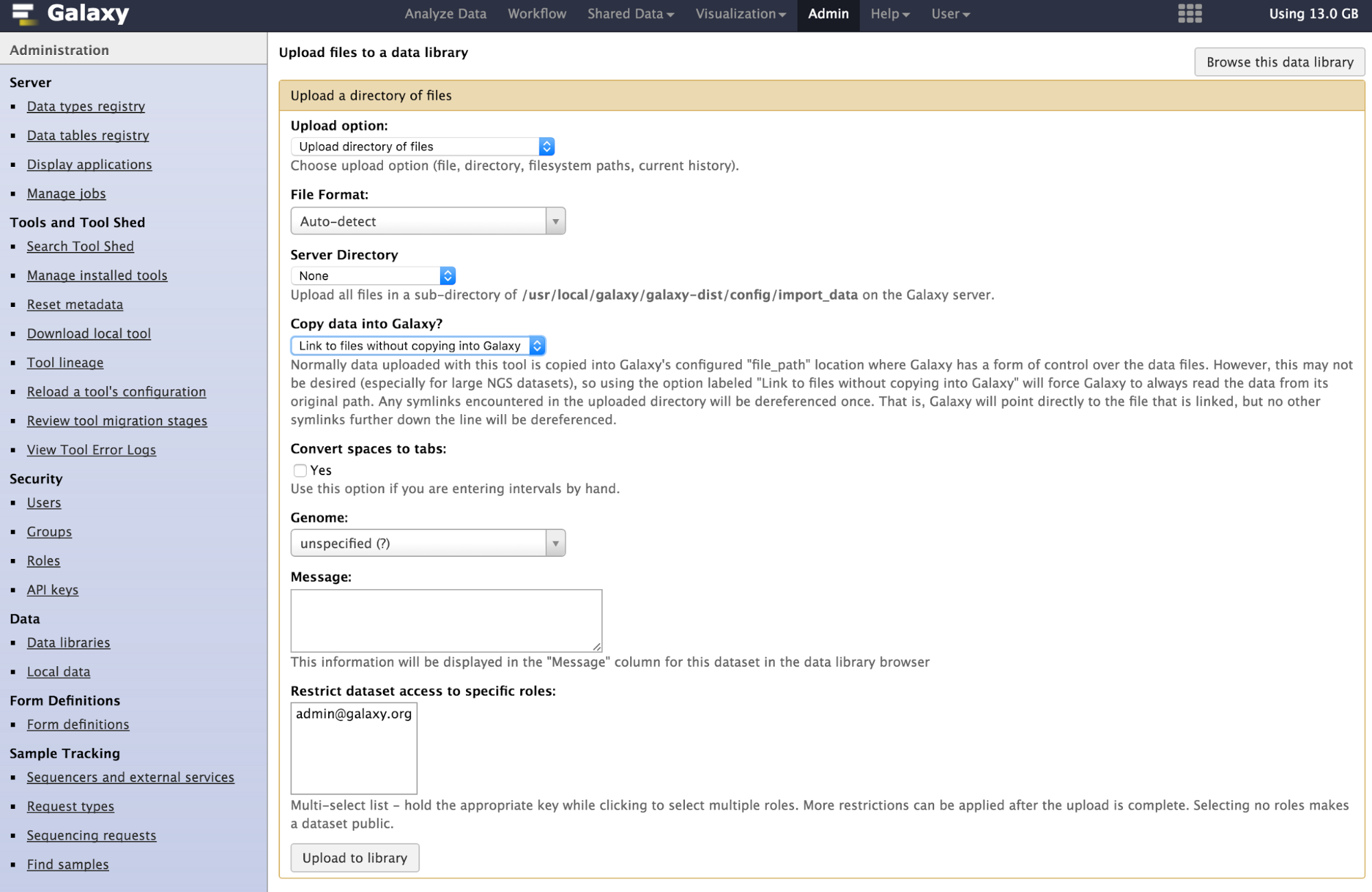
- Upload: directory of filesを選択
- File Format: fastaを選択
- Server Directory: adapter_primerを選択
- Copy Data into Galaxy?: Link to files without copying into Galaxy を選択
transcriptome_ref_fastaのデータを入れるために、もう1度、3から7まで作業する。ただし、5で入力するNameはtranscriptome_ref_fasta.
7で入力する内容は以下の通り。
- Upload options: Upload direcoty of filesを選択
- File Format: fastaを選択
- Server Directory: transcriptome_ref_fastaを選択
- Copy Data into Galaxy?: Link to files without copying into Galaxy を選択
quartz_div100_rename のデータを入れるために、もう1度、3から7まで作業する。ただし5で入力する内容は、quartz_div100_rename,7で入力する内容は以下の通り。
- Upload options: Upload direcoty of filesを選択
- File Format: fastqsangerを選択
- Server Directory: quartz_div100_renameを選択
- Copy Data into Galaxy?: Link to files without copying into Galaxy を選択
Galaxyでワークフローを実行する
つづく。