2021年7月16日午前3時頃(日本時間)に公開されたAlphaFold ver.2(以下、断りがない限り単純にAlphaFoldと書く)の論文について。
論文は https://www.nature.com/articles/s41586-021-03819-2
実装は https://github.com/deepmind/alphafold
執筆した日本語総説が出ました:AlphaFold2までのタンパク質立体構造予測の軌跡とこれから
Update Information
(2023年4月5日)
AlphaFold バージョン2.3.2以降で、alphafoldの実行コマンドにおいて、構造最適化(relax)の引数指定が--run_relaxから--models_to_relax=all/best/noneに変更されています。allはすべての構造について構造最適化を実行、bestは最も予測精度が高いと考えられるモデルのみ構造最適化、noneは行わない、を表します。
(2023年1月4日)
オリジナルのAlphaFold2で出力されるpklファイルから以下のような画像を出力する方法の記事 https://qiita.com/Ag_smith/items/5c88ffeb7a2b4eca9f71 をアップロードしました。
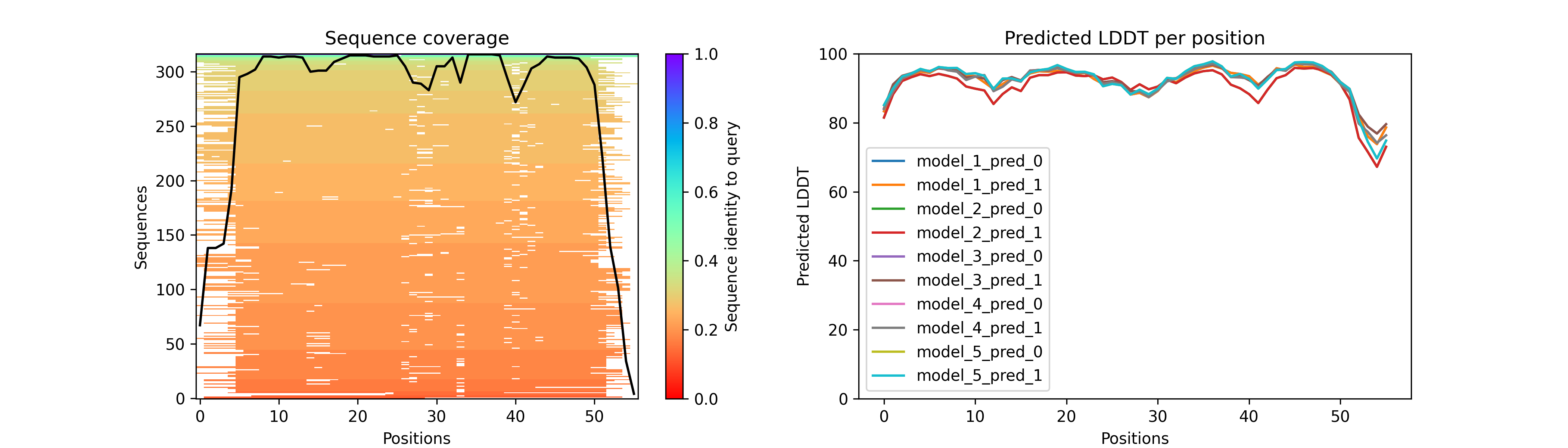
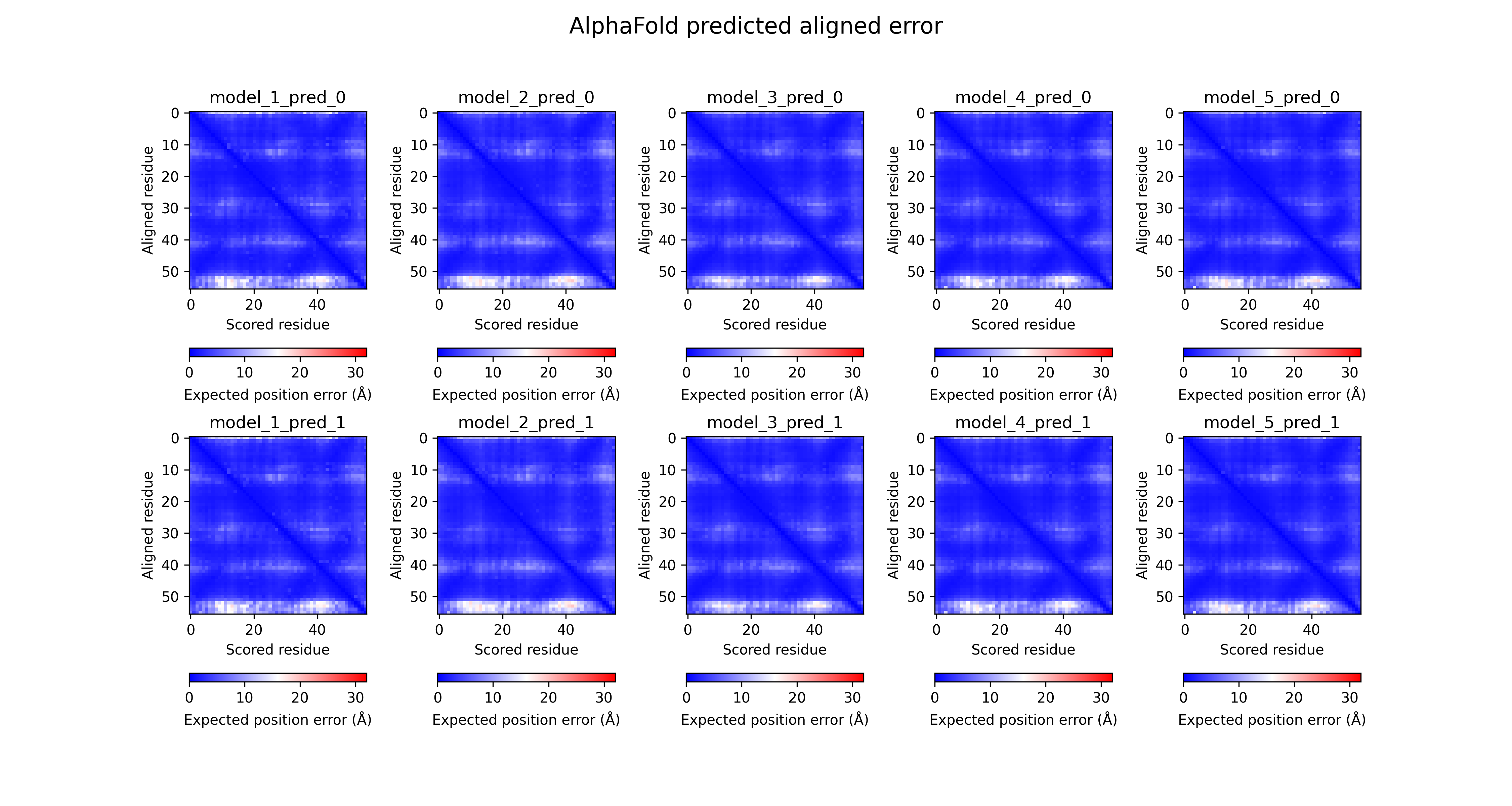
(2022年12月13日)
AlphaFold 2.3.0がリリースされました。
https://github.com/deepmind/alphafold/releases/tag/v2.3.0
特に、複合体の予測精度が2.2時代よりさらに向上したこと、複合体の予測可能限界長が伸びたことが特長のようです。
- AlphaFold複合体用の学習済みパラメータv3が利用可能(モデルアーキテクチャや学習方法は以前と同じ)
- 2021-09-30までのPDBデータを学習に使用した。以前は2018-04-30までだった。
- 様々なGPUメモリ周りの最適化を行ったことで、より長いタンパク質の予測が可能になった。
- (十分な精度がすでに出ている時に)recycleのearly stoppingを導入
- AlphaFold Colab notebook用のPython versionが3.8に。
- Relax metricsが
relax_metrics.jsonに保存されるように。
リリースノートは https://github.com/deepmind/alphafold/blob/main/docs/technical_note_v2.3.0.md
CASP15のことも含めて重要なことが色々書いてあります。
(2022年3月10日)
AlphaFold 2.2.0がリリースされました。
https://github.com/deepmind/alphafold/releases/tag/v2.2.0
-
改善された複合体予測用の学習済み重みパラメータがリリースされました。これまでの複合体予測は試験的であり、構造予測時に主鎖構造がぶつかりあって上手な予測ができていないことがありましたが、これが大きく改善されたとのことです。これに伴い、
params_model_[1-5]_multimer_v2.npzという重みパラメータが新しく配布されましたので、これを追加ダウンロードする必要があります。AlphaFoldの重みパラメータを置いているディレクトリ(/path/to/database/params)に移動し、wgetで重みパラメータの圧縮ファイルをダウンロードし、tarで解凍すればOKです。 -
--num_multimer_predictions_per_modelという引数が追加されました。デフォルトでは5に設定されています。2.1までは予測構造が5つ作られていましたが、これは5種類用意されている学習済み重みパラメータに基づくmodelが1つずつ作られて、計5個生成されるという設定になっていました。2.2からは、この5種類のmodelにつき、異なる乱数値を指定回数(デフォルトでは5)分だけ繰り返し、微妙に異なる予測構造を返すようになり、結果としてデフォルトでは25個の予測構造が生成されるようになります。これは多状態の構造を取りうるタンパク質について計算するときに便利かもしれないと示唆されていますが、その分計算時間が長くなってしまうので、これまで通り5個で十分という方は、AlphaFold 2.2の実行時に--num_multimer_predictions_per_model=1を指定してあげましょう。 -
2.1に試験的に導入されていたprokaryotic MSA pairing algorithmは、あまり効果がなかったためなくなりました。
(2022年1月28日)
AlphaFold 2.1.2がリリースされました。
https://github.com/deepmind/alphafold/releases/tag/v2.1.2
- AlphaFold2の学習済み重みパラメータが商用利用可能になりました(下記参照)。
- 実行時に
--use_gpu_relaxをオプションとしてつけることで、relax計算がGPUを用いて行われるようになり、該当処理部分が3倍以上高速化されるようになりました。デフォルトではOFFになっていますが、GPUがついているスパコン上での計算ではつけることが推奨されます。 - DockerでAlphaFold2を動かす場合、これまではrootユーザー扱いでしか動かせませんでしたが、現ユーザーで動かすことも可能になりました。
--docker_userフラグを使えばよいそうです。
(2022年1月20日)
AlphaFoldの学習済みモデルパラメータ、およびテンプレート検索時に使用しているPDB70などのデータベースのライセンスがCC BY-4.0に変更され、公式に配布されているこれらのデータベースとAlphaFoldを使って予測された立体構造は自由に商用利用可能になりました。創薬業界には嬉しいニュースです。
(2021年11月5日)
11月3日に複合体予測が可能になったAlphaFold 2.1.0がリリースされました。
https://github.com/deepmind/alphafold/releases/tag/v2.1.0
これに伴い、コード仕様や使用するデータベースが2.0時代から追加され、2.0からのアップデートに少し準備が必要になります。これについても下の方で追記します。
2.2.0系列から2.3.0へのアップデートで対応すべきこと
-
https://storage.googleapis.com/alphafold/alphafold_params_2022-12-06.tar から新しい複合体用パラメータをダウンロードして
paramsディレクトリに置く
cd /path/to/database/params
wget https://storage.googleapis.com/alphafold/alphafold_params_2022-12-06.tar
tar --extract --verbose --file=./alphafold_params_2022-12-06.tar --directory="./" --preserve-permissions
-
databaseで使う配列・構造データベースのバージョンが上がっているのでアップデートしておく。- Uniclust30データベースがuniref30データベースに名称が変更されている。scripts/download_uniref30.shで2021_03バージョンのUniRef30配列データベースがダウンロードできる。ただ今はさらに新しい2023_02バージョンが存在するのでそちらの
https://wwwuser.gwdg.de/~compbiol/uniclust/2023_02/UniRef30_2023_02_hhsuite.tar.gzを解凍して使った方がいい。 - MGnifyの配列データベースの2022_05バージョンが公式推奨となっている。ただ今はさらに新しい2023_02バージョンが存在するのでそちらの
mgy_cluster.fa.gzを解凍して使った方がいい。
- Uniclust30データベースがuniref30データベースに名称が変更されている。scripts/download_uniref30.shで2021_03バージョンのUniRef30配列データベースがダウンロードできる。ただ今はさらに新しい2023_02バージョンが存在するのでそちらの
- 依存パッケージのバージョンが上がっている。JAX, jaxlib(cuda対応)のバージョンが0.3.25、dm-haikuが0.0.9が必須。condaとpipで入れるときに注意。詳細はAlphafold本体のインストールの項へ。
-
run_alphafold.py(またはそのエイリアスのbin/alphafoldコマンド)の中の必須の配列データベース引数が--uniclust30_database_pathから--uniref30_database_pathに変わっている。上記Uniclust30データベースがuniref30になったことに対応している。地味な変更だがこれをしないと動かない。
公式のGoogle Colab版AlphaFold 2.1 Notebook
現在はDeepMindが公式に用意した、ウェブブラウザ上で動くGoogle Colab版AlphaFold 2.1 Notebookが公開されています。
https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb
こちらは2.5TBほどのデータベースを自前で用意する必要がないために、お試し感覚で使う分にはとても便利です。2.1なので公式に複合体構造予測も可能です。
しかし、注意書きあるようにこれは完全版と違って
- 配列データベースにBFDではなくreduced_bfdを使っているため、類縁配列の取得性能が落ち、珍しいタンパク質の構造予測の精度が大きくことがある(多くのホモログが多いタンパク質なら無事)。
- Google Colab無料版で適用されるGPUの仕様上、1000〜1400残基以上のタンパク質については構造予測ができません。複合体の場合は合計したアミノ酸の残基数でカウントします。課金してColab Pro版にするとグレードの高いGPUが割り当てられることが増え、この制限は少しは緩和されます(それでも2000残基程度?)。
- 複合体の性能予測については精度が落ちることが多いため、ローカル版を使ってくださいとのこと。
- Google Colab無料版は90分ほど何も操作しない状態が続くとセッションが途切れてしまい、1からやり直しになります(本当に90分かどうかは不明)。これは多量体予測の場合によくひっかかります。また12時間以上(?)使い続けて乱用していると制限にひっかかって、しばらくの間計算させてもらえなくなります。これを回避するには課金してColab Pro版にしてくださいとのこと。
ということで、複合体予測のときにはProにするかローカル版を使ったほうが良さそうです。
ColabFold
2021年7月19日頃から、Google Colab上で動かせるAlphaFold2であるColabFold を制作してくれた方々がいらっしゃいます。Martin Steinegger博士(現ソウル国立大学助教)たちHH-suiteを開発しているSöding Lab.の方々と、RoseTTAFoldの開発の1人であるSergey Ovchinnikov博士(現ハーバード大学PI)たちです。
2021年9月頃から私もColabFoldチームに加入し、様々なメンテナンスや開発を行ったことで、このソフトウェアについての論文が2022年5月に出ました
→ ColabFold: making protein folding accessible to all
使い方は簡単で、このGoogle Colabratoryのquery sequenceのところに予測したいアミノ酸配列をコピペし、上部メニューの[ランタイム]から[すべてのセルを実行]とするだけで基本的にはすべて予測が自動的に行われます。オリジナルのAlphaFoldにない特長として
- 本家よりもMSA計算が20〜30倍ほど速く、それでいて多くの場合本家AlphaFoldと同じ精度を示します。これはMSA計算部分をMartin Steinegger博士らが開発しているmmseqs2(hhblitsの後続で上位版)で計算しており、RAM 1TBほどの大容量ウェブサーバーで計算させているためです。500アミノ酸くらいのタンパク質ですと10〜20分くらいで予測が終わります。最大1000アミノ酸くらいまで予測可能です(複合体の場合は合計アミノ酸数でカウント)。AlphaFold 2.3の登場以降はアルゴリズムが改良され、ホモ多量体のときは計算可能なアミノ酸数の限界長が増えました(〜1800程度?)。
- 出力された構造の妥当性を評価するpLDDTスコアやPredicted aligned error(PAE)スコアが自動的に図示されて表示されて非常に便利。
- ColabFoldの機能を自前のGPU付きパソコン・スパコン上で動かしたい場合は私のメンテナンスしているLocalColabFoldをお使いください。
AlphaFold2を使ってみた人々の反応(7月19日)
-
多くのタンパク質については結晶構造解析の分子置換に使えるレベルで超高精度の予測結果を返してくれた。
https://twitter.com/sugargroove/status/1416959217257250818
複合体形成を行うタンパク質A,B,C...が配列データベース上にそれぞれたくさんの類縁配列を持つものならば、その数が多ければ多いほど精度が上がります。このため、データベースを豊富に持ち時間をかけて本気で類縁配列を検索する本家版の方が予測成功率が高いようです。
これはタンパク質の共進化原理に基づいた複合体対象予測として1990年代には知られていた手法のようですが、AlphaFold2の主眼である単量体(モノマー)の構造予測でも、大量の類縁配列から抽出した共進化情報を用いてどのアミノ酸ペアが構造上近位になるかを予測しています。さらなる原理についてはGoogleで「共進化 コンタクト予測」と調べてみてください。
以上のことから、類縁配列が少ない抗原・抗体間のヘテロ複合体予測は難しいんじゃないかなと思います。
-
膜タンパク質はうまく行かない(うまくいくこともある)
https://twitter.com/biochem_fan/status/1416616029912911874
https://twitter.com/ttkkmg/status/1416377949318569984
一応それらしいきれいな構造は返してくれますが、PDBに近しい構造がない場合、重要な領域は外すことが多いみたいです。
-
Intrinsic Disordered Protein (IDP)については効果なし
https://twitter.com/torusengoku/status/1416905985759711234
天然変性タンパク質については予想できません。
インストール手順(AlphaFold 2.1版)
マシン環境
十分なマシンスペックが要求されます。具体的には
- 2.5TB以上のSSD容量 (必須)
- CUDA11に対応しているNVIDIA製GPU(事実上必須)
- 大容量(32GB以上)のRAM(推奨)
OSは事実上LinuxまたはWindowsに限られます。計算速度を考えると、大容量のRAM(いわゆるメモリのこと)があればあるほど計算が速くなり、次点でRead/Write速度が速いSSDが望ましいです。おすすめ構成は
- 容量が2.5TB以上あるM.2のSSD(SATA接続でも良い)
- メモリ32GB(64GBあればなお良い)
- NVIDIA GeForce RTXの3060以上のGPU, 特に、VRAMの値が大きい3060、3090、4090がおすすめ
が備わったゲーミングデスクトップパソコン(新品ならだいたい40万円前後)での動作です。GPUはVRAMの値に大きいほど計算可能なアミノ酸配列長の限界が大きくなります(配列長の2乗に比例するそうです)。
GPUのおすすめはコスパ面でRTX3060(VRAM 12GB)で、RTX3060Ti(VRAM 8GB)のようにVRAMが小さいものはAlphaFold2には向いていません。RTX3090, 4090はVRAMが24GBありますが非常に高いです……。
AlphaFold2の構造推論処理はHH-suiteによるMSA(Multiple sequence alignment)取得が計算時間の8〜9割、Deep Learningによる学習済みモデルを用いた構造推論が1割を占めるので、速さを求めるなら大容量SSD, 大容量RAMがあるマシンを使いましょう。HH-suiteの計算時間向上についてはHH-suiteのWikiが詳しいです。
データベースの準備
AlphaFoldを実行するためには以下の配列&構造データベースが必要なのでダウンロードしておきます。
- UniRef90,
- MGnify,
- BFD,
- uniref30,
- PDB70,
- PDB (structures in the mmCIF format).
- PDB seqres – 複合体予測時にのみ必要
- UniProt – 複合体予測時にのみ必要
公式GitHubがscripts/download_all_data.sh に全部一括でダウンロードしてくれるスクリプトを置いてくれているので、それを使えば全部いい感じにやってくれます。これには8〜12時間かかるとされていますが日本からだともっとかかります。実際にはこのスクリプトを実行する前にいくつか変更を加えておいた方がこの辺の準備を早く行えます(後述)。
このスクリプトを使う場合は先に強力なダウンロードコマンドのrsyncとaria2(aria2c)をインストールしておく必要があります。インストールは
# for Debian OS (Ubuntu)
sudo apt -y install rsync aria2
# for RedHat OS (CentOS or Fedora)
sudo yum -y install epel-release && sudo yum -y install rsync aria2
でできます。
次に先述したデータベースのインストールディレクトリ/data/moriwaki/databaseを用いて、そのディレクトリにデータベースをダウンロードしていきます(が、下記の内容を含めてこの項全体をよく読んでから実行することをおすすめします)
DOWNLOAD_DIR="/data/moriwaki/database"
scripts/download_all_data.sh ${DOWNLOAD_DIR}
とすることで、その指定したディレクトリにデータベースを順々にダウンロードしてくれます。
このスクリプトの実態は同じディレクトリにある他7つ9つのダウンロード実行スクリプト
-
download_alphafold_params.sh(AlphaFold 2.1追加時に更新された) -
download_bfd.sh- (
download_small_bfd.sh) : BFDと択一。small_bfdはお試し用。
- (
download_mgnify.shdownload_uniref90.sh-
download_uniref30.sh(2.2以前はdownload_uniclust30.shという名前だった) download_pdb70.shdownload_pdb_mmcif.sh-
download_uniprot.sh(AlphaFold 2.1から追加) -
download_pdb_seqres.sh(AlphaFold 2.1から追加)
これらを順番に実行するものとなっており、これらのスクリプトを並列で実行した方が早いです。並列で実行したい場合は
DOWNLOAD_DIR="/data/moriwaki/database"
nohup scripts/download_alphafold_params.sh ${DOWNLOAD_DIR} > dl_params.out &
nohup scripts/download_bfd.sh ${DOWNLOAD_DIR} > dl_bfd.out &
...(以下略)
みたいな感じでバックグラウンド処理させると良い。dl_xxx.outファイルにそのダウンロードの記録が残るような仕組みになっています。また、各コマンドの先頭にnohupをつけて開始させると、ssh接続を閉じたりターミナルを閉じたりしても処理が続行されるので便利です。もしその処理を止めたい場合はps auxでプロセス番号(PID)を調べてからkill -9 <PID>とします。
以下は上記コマンドを実行する前に知っておくと便利なTIPS。
download_alphafold_params.sh(AlphaFold 2.3で更新が必要)
学習済みのalphafoldのパラメータたちをダウンロードします。注意点として、AlphaFoldのコードはすべてApache 2.0 Licenseであるため完全に自由に改変していいし商用利用も可能ですが、これでダウンロードされるパラメータファイルはCC BY-NC 4.0 Licenseなので、使用時には適切なクレジット表示をしなければならず、営利目的での利用は禁止されています。
(2022年1月20日:このパラメータのライセンスがCC BY-4.0、つまり商用利用可能に変更されました。)
download_bfd.sh
AlphaFoldの構造予測プロセスの初期段階で用いるHH-suite ver.3.3で、クエリ配列に対するリモートホモログ配列を取得するときに使う追加データベースのBFDをダウンロードする。https://bfd.mmseqs.com/ からダウンロード可能。下記のuniclust30データベースに加えてUniprot/Tremble, Swiss-protといった完全な配列だけでなく、メタゲノム配列を加えてさらに配列空間を増強することで共進化情報の精度を大幅に上げることに成功したという2017年の論文以降、その要因でHH-suiteチームも公式にメタゲノム配列を加えたデータベースBFDを提供し定期的に更新して配信してくれています。
しかし、データサイズが非常に大きい。2.2TBくらいある。 これが理由で大容量HDD(可能ならSSDの方が相当高速化される)が要求されます。このデータベースは上記の通り使わなくても原理上は動かすことは可能ですが、あった方が大変よい。
解凍後の当該ディレクトリはこのようなファイル構成になります。ただし、初期設定ではパーミッションが600になっているため、共用計算機上で使う場合はchmod 644 *としてreadableにしておきましょう。
[/data/moriwaki/database/bfd] $ ls -lt
-rw-r--r-- 1 root root 1688518403 Mar 5 2019 bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_cs219.ffindex
-rw-r--r-- 1 root root 16814534465 Mar 5 2019 bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_cs219.ffdata
-rw-r--r-- 1 root root 1555724298013 Mar 4 2019 bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_a3m.ffdata
-rw-r--r-- 1 root root 326882934659 Mar 3 2019 bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_hhm.ffdata
-rw-r--r-- 1 root root 129631317 Mar 3 2019 bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_hhm.ffindex
-rw-r--r-- 1 root root 1817298807 Mar 3 2019 bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_a3m.ffindex
download_uniref30.sh
2.2以前はdownload_uniclust30.shという名前でした。AlphaFoldの構造予測プロセスの初期段階で用いるHH-suite ver.3.3で、クエリ配列に対するリモートホモログ配列を取得するときに必須のデータベースです。現在は http://wwwuser.gwdg.de/~compbiol/uniclust/2023_02/ に置かれている最新版の UniRef30_2023_02_hhsuite.tar.gz を用いることが有効ですので、download_uniclust30.shを書き換えてから使ってください。さらに、初期設定ではパーミッションが600になっているため、共用計算機上で使う場合はchmod 644 *としてreadableにしておきましょう。
-rw-r--r-- 1 moriwaki staffs 9051302414 May 21 2023 UniRef30_2023_02_cs219.ffdata
-rw-r--r-- 1 moriwaki staffs 889852336 May 21 2023 UniRef30_2023_02_cs219.ffindex
-rw-r--r-- 1 moriwaki staffs 218410841490 May 21 2023 UniRef30_2023_02_a3m.ffdata
-rw-r--r-- 1 moriwaki staffs 986967200 May 21 2023 UniRef30_2023_02_a3m.ffindex
-rw-r--r-- 1 moriwaki staffs 51112196683 May 21 2023 UniRef30_2023_02_hhm.ffdata
-rw-r--r-- 1 moriwaki staffs 28407605 May 21 2023 UniRef30_2023_02_hhm.ffindex
-rw-r--r-- 1 moriwaki staffs 379 May 21 2023 UniRef30_2023_02.md5sums
※パーミッションが-rw-r--r--のようにreadableとなっていることを確認しましょう。
ちなみに2021_03版はいくつかの配列の予測の時にエラーを起こすことが知られています。使わないようにしましょう。
download_mgnify.sh
メタゲノムデータベースの1つであるMGnifyの配列データセットのダウンロード。デフォルトでは https://storage.googleapis.com/alphafold-databases/v2.3/mgy_clusters_2022_05.fa.gz のウェブサイトから圧縮されたmgy_clusters_2022_05.fa.gzのFASTA形式アミノ酸配列データセットをダウンロードしてくるので、これを解凍して配置しているだけです。
これは2022年5月時点のものですが、実は https://ftp.ebi.ac.uk/pub/databases/metagenomics/peptide_database に行くと最新の2023年2月版のものがおいてあるので、2022_05版よりも最新版をダウンロードして利用するように設定しておくと、AlphaFoldの仕組みを考えると構造予測精度の上昇につながることが期待されます。
download_uniref90.sh
タンパク質のアミノ酸配列と機能情報を高品質で包括的に提供することを目指すUniprotデータベースに置かれてあるUniProt Reference Clusters (UniRef)の配列。UniRef100に含まれるアミノ酸配列をクラスタリングすることで構築されており、各クラスタは最長の配列(シード配列)に対して90%以上の相同性を持ち、かつ80%以上のオーバーラップを持つ配列で構成されています。
UniRef90 is built by clustering UniRef100 sequences such that each cluster is composed of sequences that have at least 90% sequence identity to, and 80% overlap with, the longest sequence (a.k.a. seed sequence).
解凍されたuniref90.fastaファイルの中には大量のアミノ酸配列がFASTAフォーマットでテキスト形式で書かれており、これとUniclust30 + BFDデータベースとHH-suiteソフトウェアを用いることで、uniref90.fastaファイルの中からクエリ配列に対するリモートホモログを検出します。
(2023年6月2日更新) スイスのExpasyのミラーデータベースからダウンロードするようにスクリプトを書き換える、または手動でここからダウンロードしたほうが速いです。
※日本のDDBJでミラーリングされているuniref90の配列データは現在壊れているようです。
download_pdb70.sh
HH-Suiteが提供するデータベースの1つで、Protein Data Bankに登録されている構造の配列をクラスタリングしたもの。
http://wwwuser.gwdg.de/~compbiol/data/hhsuite/databases/hhsuite_dbs/ から最新版をダウンロード可能。
元はHH-Suiteのチームが2005年に作成した構造予測プログラムHHPredのために作られているデータベース。テンプレートベースの構造予測としては非常に高速で強力です。AlphaFoldはこれと似たアルゴリズムでテンプレートとして計算されたタンパク質立体構造を構造予測の出発点としているみたいです。
解凍後はこんな感じ。初期設定ではパーミッションが600になっているため、共用計算機上で使う場合はchmod 644 *としてreadableにしておきましょう。
[/data/moriwaki/database/pdb70] $ ls -lt
-rw-r--r-- 1 moriwaki moriwaki 410 Jul 8 23:52 md5sum
-rw-r--r-- 1 moriwaki moriwaki 3875841099 Jul 8 23:47 pdb70_hhm.ffdata
-rw-r--r-- 1 moriwaki moriwaki 2066799 Jul 8 23:47 pdb70_hhm.ffindex
-rw-r--r-- 1 moriwaki moriwaki 63005744825 Jul 8 23:46 pdb70_a3m.ffdata
-rw-r--r-- 1 moriwaki moriwaki 2245399 Jul 8 23:46 pdb70_a3m.ffindex
-rw-r--r-- 1 moriwaki moriwaki 24942439 Jul 8 23:08 pdb70_cs219.ffdata
-rw-r--r-- 1 moriwaki moriwaki 1715919 Jul 8 23:08 pdb70_cs219.ffindex
-rw-r--r-- 1 moriwaki moriwaki 8204591 Jul 7 10:01 pdb70_clu.tsv
-rw-r--r-- 1 moriwaki moriwaki 23827536 Jul 7 10:00 pdb_filter.dat
AlphaFoldのデータベースのタイムスタンプは2020-05-13となっていますが、特にこだわりがない場合は最新のものを使う方が良いでしょう。
追記: http://ftp.tuebingen.mpg.de/pub/protevo/toolkit/databases/hhsuite_dbs/ ここに最新のPDB70データベースが置かれるようです。
download_pdb_mmcif.sh (2021年11月5日追記)
公式の説明によれば
Remove /pdb_mmcif. It is needed to have PDB SeqRes and PDB from exactly the same date. Failure to do this step will result in potential errors when searching for templates when running AlphaFold-Multimer.
とあるように、後述のpdb_seqres.txt.gzのファイルの更新日とpdb_mmcifのデータベースの更新日を同じものにしておく必要があり、こうしないと複合体予測時に限ってエラーが起きるかもしれない、とのことです。
これはProtein Data Bank(PDB)のmmCIFフォーマットの構造データをまるごとローカルにミラーリングするスクリプトです。
しかし日本から利用する場合はrsyncする対象のウェブサイトを日本のミラーサーバーに変更してから実行した方が圧倒的に速い。そこで44〜45行目の部分を
- rsync --recursive --links --perms --times --compress --info=progress2 --delete --port=33444 \
- rsync.rcsb.org::ftp_data/structures/divided/mmCIF/ \
+ rsync --recursive --links --perms --times --compress --info=progress2 --delete \
+ ftp.pdbj.org::ftp_data/structures/divided/mmCIF/ \
このように書き換え、改めて
scripts/download_pdb_mmcif.sh /path/to/installdir
とすればおよそ1時間くらいでデータベース作成が終わります。
ちなみに
Running rsync to fetch all mmCIF files (note that the rsync progress estimate might be inaccurate)...
Welcome to PDB Archive Rsync service!
54,468,303,486 100% 64.56MB/s 0:13:24 (xfr#183793, to-chk=0/199277)
Unzipping all mmCIF files...
このxfrの数字はRCSB PDBのトップに表示されているデータ数199277と一致します(※2022年12月当時)。PDBの構造データは週に一度、金曜日あたりかな、その頃にアップデートされます。
download_uniprot.sh (AlphaFold 2.1以降で必要)
セットアップ方法は公式マニュアルに書いてあるように
scripts/download_uniprot.sh /path/to/installdir
です。ここで/path/to/installdirは任意のディレクトリ名です。今のディレクトリで良ければ.とだけ入力します。
やっていることはUniProtという配列データベースに存在するアミノ酸配列ファイルの全データuniprot_sprot.fasta.gzとuniprot_trembl.fasta.gzをダウンロードし、解凍して連結してuniprot.fastaというファイルを作成することです。現時点では
-rw-r--r-- 1 root root 120546547214 Jun 2 14:01 uniprot.fasta
のファイルサイズです(2023_02 ver.)。
つまり実体的には
DOWNLOAD_DIR="/path/to/installdir"
ROOT_DIR="${DOWNLOAD_DIR}/uniprot"
cd $DOWNLOAD_DIR
rm -rf $ROOT_DIR
mkdir $ROOT_DIR && cd $ROOT_DIR
wget https://ftp.expasy.org/databases/uniprot/current_release/knowledgebase/complete/uniprot_trembl.fasta.gz -O uniprot_trembl.fasta.gz --no-check-certificate
wget https://ftp.expasy.org/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz -O uniprot_sprot.fasta.gz -O uniprot_sprot.fasta.gz --no-check-certificate
# この2つのファイルの解凍
gunzip uniprot_trembl.fasta.gz
gunzip uniprot_sprot.fasta.gz
# Concatenate TrEMBL and SwissProt, rename to uniprot and clean up.
cat uniprot_sprot.fasta >> uniprot_trembl.fasta
mv uniprot_trembl.fasta uniprot.fasta
rm uniprot_sprot.fasta
を実行していることと等価です。
pdb_seqresデータベース(AlphaFold 2.1以降で必要)
PDBに登録されているタンパク質のアミノ酸配列のデータベースです(FASTA形式)。例えばHisタグとかが入っていたまま結晶化されたタンパク質など、そのへんの情報も含まれています。
これについてもセットアップ方法は公式マニュアルに書いてあるように
scripts/download_uniprot.sh /path/to/installdir
で終わる……のですが、実際にはサーバーの位置の都合上非常にダウンロードが遅いため、pdb_mmcifのときと同様にこのダウンロードスクリプトの34行目を
- SOURCE_URL="ftp://ftp.wwpdb.org/pub/pdb/derived_data/pdb_seqres.txt"
+ SOURCE_URL="https://ftp.pdbj.org/pub/pdb/derived_data/pdb_seqres.txt.gz"
のように日本のPDBjのサーバーからダウンロードするように書き換えてあげましょう。このように変更することを強く推奨します。これで一瞬でダウンロードが終わります。
終わったらこれを解凍して
cd /path/to/installdir/pdb_seqres
gunzip pdb_seqres.txt.gz
こうなっていればOK
-rw-r--r-- 1 root root 217994548 Nov 4 13:37 pdb_seqres.txt
実体的には
DOWNLOAD_DIR="/path/to/installdir"
ROOT_DIR="${DOWNLOAD_DIR}/pdb_seqres"
cd $DOWNLOAD_DIR
rm -rf $ROOT_DIR
mkdir -p pdb_seqres
wget https://ftp.pdbj.org/pub/pdb/derived_data/pdb_seqres.txt.gz
gunzip pdb_seqres.txt.gz
mv pdb_seqres.txt $ROOT_DIR
を実行していることと等価です。
最終的には、databaseディレクトリ以下はこんなファイル構成になっているはずです(2022年12月のver.2.2.0以降)。
database
├── bfd
│ ├── bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_a3m.ffdata
│ ├── bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_a3m.ffindex
│ ├── bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_cs219.ffdata
│ ├── bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_cs219.ffindex
│ ├── bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_hhm.ffdata
│ └── bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_hhm.ffindex
├── mgnify
│ └── mgy_clusters_2022_05.fa #またはmgy_clusters.fa (2023_02版)
├── params
│ ├── LICENSE
│ ├── params_model_1_multimer.npz # AlphaFold 2.1のみ必要。現在は不要。
│ ├── params_model_1_multimer_v2.npz # AlphaFold 2.2のみ必要。現在は不要。
│ ├── params_model_1_multimer_v3.npz
│ ├── params_model_1.npz
│ ├── params_model_1_ptm.npz
│ ├── params_model_2_multimer.npz # AlphaFold 2.1のみ必要。現在は不要。
│ ├── params_model_2_multimer_v2.npz # AlphaFold 2.2のみ必要。現在は不要。
│ ├── params_model_2_multimer_v3.npz
│ ├── params_model_2.npz
│ ├── params_model_2_ptm.npz
│ ├── params_model_3_multimer.npz # AlphaFold 2.1のみ必要。現在は不要。
│ ├── params_model_3_multimer_v2.npz # AlphaFold 2.2のみ必要。現在は不要。
│ ├── params_model_3_multimer_v3.npz
│ ├── params_model_3.npz
│ ├── params_model_3_ptm.npz
│ ├── params_model_4_multimer.npz # AlphaFold 2.1のみ必要。現在は不要。
│ ├── params_model_4_multimer_v2.npz # AlphaFold 2.2のみ必要。現在は不要。
│ ├── params_model_4_multimer_v3.npz
│ ├── params_model_4.npz
│ ├── params_model_4_ptm.npz
│ ├── params_model_5_multimer.npz # AlphaFold 2.1のみ必要。現在は不要。
│ ├── params_model_5_multimer_v2.npz # AlphaFold 2.2のみ必要。現在は不要。
│ ├── params_model_5_multimer_v3.npz
│ ├── params_model_5.npz
│ └── params_model_5_ptm.npz
├── pdb70
│ ├── md5sum
│ ├── pdb70_a3m.ffdata
│ ├── pdb70_a3m.ffindex
│ ├── pdb70_clu.tsv
│ ├── pdb70_cs219.ffdata
│ ├── pdb70_cs219.ffindex
│ ├── pdb70_hhm.ffdata
│ ├── pdb70_hhm.ffindex
│ └── pdb_filter.dat
├── pdb_mmcif
│ ├── mmcif_files
│ │ ├── 100d.cif
│ │ ├── 101d.cif
│ │ ├── .........(以下たくさんのcifファイル)
│ └── obsolete.dat
├── pdb_seqres
│ └── pdb_seqres.txt
├── small_bfd (※bfdディレクトリがあれば不要)
│ └── bfd-first_non_consensus_sequences.fasta
├── uniprot
│ └── uniprot.fasta
├── uniref30/UniRef30_2023_02
│ ├── UniRef30_2023_02_cs219.ffdata
│ ├── UniRef30_2023_02_cs219.ffindex
│ ├── UniRef30_2023_02_a3m.ffdata
│ ├── UniRef30_2023_02_a3m.ffindex
│ ├── UniRef30_2023_02_hhm.ffdata
│ ├── UniRef30_2023_02_hhm.ffindex
│ └── UniRef30_2023_02.md5sums
└── uniref90
└── uniref90.fasta
AlphaFold インストールのやり方1: Dockerを使う場合
※2021年8月以降、Dockerを使ったインストール方法の仕様変更についていけていないため、今はやり方2の方を推奨します。可能ならそのうち修正します。
aptを使ったインストール
Ubuntu20.04の場合、以下の手順で導入します。
# aptのアップデート
sudo apt -y update
# aptがHTTPS経由でパッケージを使用できるようにするいくつかの必要条件パッケージをインストール
sudo apt -y install apt-transport-https ca-certificates curl software-properties-common
# 公式DockerリポジトリのGPGキーをシステムに追加
curl -fsSL https://download.docker.com/linux/ubuntu/gpg | sudo apt-key add -
sudo add-apt-repository "deb [arch=amd64] https://download.docker.com/linux/ubuntu $(lsb_release -cs) stable"
# 追加されたリポジトリからDockerパッケージでパッケージデータベースを更新
sudo apt -y update
# docker-ceのインストール
sudo apt-get install -y docker-ce
以上でDockerがインストールされます。dockerを確認します。
$ docker version
Client: Docker Engine - Community
Version: 20.10.7
API version: 1.41
Go version: go1.13.15
Git commit: f0df350
Built: Wed Jun 2 11:56:38 2021
OS/Arch: linux/amd64
Context: default
Experimental: true
Got permission denied while trying to connect to the Docker daemon socket at unix:///var/run/docker.sock: Get http://%2Fvar%2Frun%2Fdocker.sock/v1.24/version: dial unix /var/run/docker.sock: connect: permission denied
Dockerがインストールされ、デーモンが起動し、プロセスがプート時に起動できるようになりました。実行されていることを確認します。
$ systemctl status docker
● docker.service - Docker Application Container Engine
Loaded: loaded (/lib/systemd/system/docker.service; enabled; vendor preset: enabled)
Active: active (running) since Fri 2021-07-16 15:52:09 JST; 9min ago
TriggeredBy: ● docker.socket
Docs: https://docs.docker.com
Main PID: 298145 (dockerd)
Tasks: 32
Memory: 50.7M
CGroup: /system.slice/docker.service
└─298145 /usr/bin/dockerd -H fd:// --containerd=/run/containerd/containerd.sock
Active: active (running)になっていれば成功。正しく起動できていない場合はここが赤色で表示されます。
Dockerを非rootユーザーでも使えるようにする
rootでなく一般ユーザでdockerを実行する場合には、Linuxの場合dockerグループに所属している必要があります。このため任意のユーザーを管理者権限でひとりひとりdocker groupに入れてあげます。例えばmoriwakiユーザーをdockerグループに入れたい場合は
sudo usermod -aG docker moriwaki
と実行する必要があります。
ここまで終わったら一度ログアウトし、再びログインした後にコマンドdocker run hello-worldを実行します。
$ docker run hello-world
Unable to find image 'hello-world:latest' locally
latest: Pulling from library/hello-world
b8dfde127a29: Pull complete
Digest: sha256:df5f5184104426b65967e016ff2ac0bfcd44ad7899ca3bbcf8e44e4461491a9e
Status: Downloaded newer image for hello-world:latest
Hello from Docker!
This message shows that your installation appears to be working correctly.
このようなメッセージが表示されていれば準備OKです。
NVIDIA Container Toolkitのインストール
Dockerをインストールし終わったあとで、公式の記述 https://docs.nvidia.com/datacenter/cloud-native/container-toolkit/install-guide.html を元にインストールします。
distribution=$(. /etc/os-release;echo $ID$VERSION_ID) \
&& curl -s -L https://nvidia.github.io/nvidia-docker/gpgkey | sudo apt-key add - \
&& curl -s -L https://nvidia.github.io/nvidia-docker/$distribution/nvidia-docker.list | sudo tee /etc/apt/sources.list.d/nvidia-docker.list
この後、nvidia-docker2 packageをインストールします。
sudo apt-get update
sudo apt-get install -y nvidia-docker2
インストールを完了するために、dockerのデーモンを再起動します。
sudo systemctl restart docker
ここで正しく動作しているかどうかを以下のコマンドで確認します。
sudo docker run --rm --gpus all nvidia/cuda:11.0-base nvidia-smi
nvidia-smiコマンドを実行したときのように、GPU情報が表示されれば成功。こんな感じの画面。
$ docker run --rm --gpus all nvidia/cuda:11.0-base nvidia-smi
Fri Jul 16 08:24:07 2021
+-----------------------------------------------------------------------------+
| NVIDIA-SMI 460.84 Driver Version: 460.84 CUDA Version: 11.2 |
|-------------------------------+----------------------+----------------------+
| GPU Name Persistence-M| Bus-Id Disp.A | Volatile Uncorr. ECC |
| Fan Temp Perf Pwr:Usage/Cap| Memory-Usage | GPU-Util Compute M. |
| | | MIG M. |
|===============================+======================+======================|
| 0 GeForce RTX 3090 Off | 00000000:0D:00.0 Off | N/A |
| 54% 35C P0 26W / 350W | 0MiB / 24265MiB | 0% Default |
| | | N/A |
+-------------------------------+----------------------+----------------------+
+-----------------------------------------------------------------------------+
| Processes: |
| GPU GI CI PID Type Process name GPU Memory |
| ID ID Usage |
|=============================================================================|
| No running processes found |
+-----------------------------------------------------------------------------+
エラーだとこんな感じで表示されます。
docker: Error response from daemon: could not select device driver "" with capabilities: [[gpu]].
Alphafoldのリポジトリのクローン
GitHubからAlphaFoldをダウンロードしてきます。
git clone https://github.com/deepmind/alphafold.git
cd alphafold
run_dockerファイルの編集
リポジトリをクローンしたらさっそくいくつかのパラメータを変えておきます。docker/run_docker.pyファイルを開いて以下の部分を編集します。
#### USER CONFIGURATION ####
# Set to target of scripts/download_all_databases.sh
DOWNLOAD_DIR = 'SET ME'
# Name of the AlphaFold Docker image.
docker_image_name = 'alphafold'
# Path to a directory that will store the results.
output_dir = '/tmp/alphafold'
これはユーザーそれぞれが好きなように設定しておきます。例えば私はこうしました。
#### USER CONFIGURATION ####
# Set to target of scripts/download_all_databases.sh
DOWNLOAD_DIR = '/data/moriwaki/database'
# Name of the AlphaFold Docker image.
docker_image_name = 'alphafold'
# Path to a directory that will store the results.
output_dir = '/data/moriwaki/alphafold_out'
ここで指定したDOWNLOAD_DIR変数のディレクトリは、後のデータベースの準備のときのディレクトリ名と同一にします。
また、ここで設定したoutput_dirは存在していないとAlphaFold2の動作時にエラーが起きるので、ディレクトリを作成しておきます(重要)。
$ mkdir -p /data/moriwaki/alphafold_out
上のスクリプトで設定したアウトプット用ディレクトリが作成されました。
AlphaFold実行の手順
最も簡単な方法はdocker/run_docker.pyを使ってDocker環境で実行することです。
-
docker/run_docker.pyファイルの中にあるDOWNLOAD_DIRという文字を、自身の環境で設置したデータベースのディレクトリのPATHに書き換えます。例えば/data/moriwaki/databaseみたいな感じ。
# Set to target of scripts/download_all_databases.sh
- DOWNLOAD_DIR = 'SET ME'
+ DOWNLOAD_DIR = '/data/moriwaki/database'
-
Dockerイメージを構築します。
docker build -f docker/Dockerfile -t alphafold .もしbuildした後にDockerfileの設定をし忘れていていることに気付いて設定を変更する必要があった場合は、
docker imagesでalphafold2のIMAGE IDを取得後、docker rmi <IMAGE ID> -fとしてそのイメージを削除してからbuildしなおしましょう。詳しくはdockerのimageの削除などでググってください。 -
run_docker.pyの実行に必要なPythonパッケージをインストールします。(場合によってはvirtualenvなどで仮想環境を作ってシステムのPython環境とコンフリクトしないようにしておくのも手でしょう。)pip3 install -r docker/requirements.txt -
構造予測したいアミノ酸配列が入ったFASTAフォーマットのファイルを作成し、そのファイルへのPATHを指定して
run_docker.pyスクリプトを実行します。もし予測したい構造がすでにPDBに登録されており、それをテンプレートとして用いることを避けたい場合は、max_template_dateというオプションを指定することで、それ以前のPDBデータ公開日の構造をもとに構造予測することができます。例えばCASP14のターゲットのひとつT1050についてはpython3 docker/run_docker.py --fasta_paths=/path/to/T1050.fasta --max_template_date=2020-05-14またデフォルトではAlphaFoldはすべての取得可能なGPUデバイスを利用して計算しようとします。一部だけ使いたい場合は、コンマで区切られたGPUのUUIDまたはindexのリストを指定して
--gpu_devicesフラグに入力します。詳細はGPU enumerationを読んでください。
AlphaFold インストールのやり方2: Dockerを使わない場合
研究室のワークステーションやスーパーコンピュータなど、多人数で共有するマシンの上で動かしたい場合はこっちのやり方が良いかも。
NVIDIA Cuda compiler driverとNVIDIA Driverのインストール&動作確認
Linux, WindowsでNVIDIA GPUを積んでいるマシンの場合、GPUを使ってディープラーニングの計算を大幅に加速させるのに必要なドライバ(※適当な説明)。
インストール手順はそのマシンや時代に応じて少しずつやり方が変わってしまうので、正確にはNVIDIA公式のCUDAインストール手順を読んでCUDA11.8以上のドライバをインストールしましょう。特に、最新のRTX4000番台を使う場合はCUDA 11.8以上 でないと動作しません。(参考: https://en.wikipedia.org/wiki/CUDA#GPUs_supported )
インストールが完了して、ターミナルからnvcc --versionと打ったときに表示されるバージョンが11.8または12以上であればOKです。
$ nvcc --version
nvcc: NVIDIA (R) Cuda compiler driver
Copyright (c) 2005-2022 NVIDIA Corporation
Built on Wed_Sep_21_10:33:58_PDT_2022
Cuda compilation tools, release 11.8, V11.8.89
Build cuda_11.8.r11.8/compiler.31833905_0
GPUの計算を行うためには、NVIDIA Driverをインストールしておくことも必要です。cuda driverと一緒に入ることもありますが、https://www.nvidia.co.jp/Download/index.aspx?lang=jp も参考にして入れてみましょう。
インストール後、次のコマンドを打ちます。
$ nvidia-smi -L
GPU 0: NVIDIA GeForce RTX 3090 (UUID: GPU-ac6xxxxxx...........)
nvidia-smi -Lとしたときに、マシンのGPU情報が表示されれば成功です。もしNVIDIA-SMI has failed because it couldn't communicate with the NVIDIA driver. Make sure that the latest NVIDIA driver is installed and running.と現れた場合は、AlphaFoldは動作しません。マシンを再起動するか、再度インストールし直すか、設定を見直してください。
AlphaFold本体のインストール
やや上級者向けですが以下のコマンドで1行目のINSTALLDIRの部分をお好みで変えるだけであとはコピペでインストールが完了します。
※ 2022年12月14日、version 2.3.0向けに修正
※ 2023年6月14日、version 2.3.2向けに修正。OpenMMのバージョン8.0.0を使うよう修正。
※ 2023年6月15日、conda cudatoolkitが11.8以上だと不安定になることがあるようで11.6.0に変更、OpenMMのバージョンを合わせて7.7.0に修正。
## インストール先のディレクトリを適当に指定
INSTALLDIR=/home/moriwaki/apps/
## ここまで設定事項、以下は変更の必要なし
# INSTALLDIR以下にalphafoldリポジトリをGitHubからダウンロード
mkdir -p ${INSTALLDIR}
cd ${INSTALLDIR}
git clone https://github.com/deepmind/alphafold.git
cd alphafold
ALPHAFOLDDIR=${INSTALLDIR}/alphafold
# stereo_chemical_props.txtのダウンロード。動作に必須。
wget -q -P ${ALPHAFOLDDIR}/alphafold/common/ https://git.scicore.unibas.ch/schwede/openstructure/-/raw/7102c63615b64735c4941278d92b554ec94415f8/modules/mol/alg/src/stereo_chemical_props.txt --no-check-certificate
# Alphafoldの動作に必要な外部Pythonライブラリを、Miniconda仮想環境"alphafold-conda"を構築しながらその中にインストールする
cd ${ALPHAFOLDDIR}
wget -q -P . https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash ./Miniconda3-latest-Linux-x86_64.sh -b -p ${ALPHAFOLDDIR}/conda
rm Miniconda3-latest-Linux-x86_64.sh
. "${ALPHAFOLDDIR}/conda/etc/profile.d/conda.sh"
export PATH="${ALPHAFOLDDIR}/conda/condabin:${PATH}"
conda create -p ${ALPHAFOLDDIR}/alphafold-conda python=3.9 -y
conda activate ${ALPHAFOLDDIR}/alphafold-conda
conda update -n base conda -y
conda install -y -c conda-forge cudatoolkit==11.6.0 openmm==7.7.0 pdbfixer cudnn=8.2.1.32 typing-extensions
conda install -y -c bioconda hmmer==3.3.2 hhsuite==3.3.0 kalign2=2.04
pip install pandas absl-py==1.0.0 biopython==1.79 chex==0.0.7 dm-haiku==0.0.9 dm-tree==0.1.6 immutabledict==2.0.0 jax==0.3.25 ml-collections==0.1.0 numpy==1.21.6 scipy==1.7.3 tensorflow-cpu==2.11.0 protobuf==3.19.6 pandas==1.3.4
pip install https://storage.googleapis.com/jax-releases/cuda11/jaxlib-0.3.25+cuda11.cudnn82-cp39-cp39-manylinux2014_x86_64.whl
pip install --upgrade jax==0.3.25
# 仮想環境構築ここまで。
# 最後にOpenMM 7.5.1へのパッチを当てて構造最適化(relax)が正しく動作するようにする。
# いまはOpenMM 7.7.0依存になったのでこのバグは解消され、この操作は不要になった
# (cd ${ALPHAFOLDDIR}/alphafold-conda/lib/python3.9/site-packages/ && patch -p0 < ${ALPHAFOLDDIR}/docker/openmm.patch)
簡単な動作のために、生成されたalphafoldディレクトリの中に入ってbinディレクトリを作成し、以下のalphafoldコマンドを作成しておきます。
(2022年8月17日追記)
上記のalphafoldのシェルスクリプトファイルは chmod +x alphafoldで実行可能にしておく必要があります 。そしてインストールしたalphafoldディレクトリの中身は次のような構成になります。
${INSTALLDIR}/alphafold
├── alphafold
├── alphafold-conda
├── bin
│ └── alphafold #これが上記のalphafold shell script
├── conda
├── CONTRIBUTING.md
├── docker
├── imgs
├── LICENSE
├── notebooks
├── README.md
├── requirements.txt
├── run_alphafold.py
├── run_alphafold_test.py
├── scripts
└── setup.py
(追記ここまで)
これを設置してここにPATHを通せば共用計算機上で誰からでもalphafoldコマンドを呼び出すことができます。ユーザーは
# /path/toは${INSTALLDIR}で入力した場所の絶対パス
export PATH="/path/to/alphafold/bin:$PATH"
# Specify a fasta file whose structure you want to predict.
FASTAFILE="1bjp.fasta"
# Specify a folder to save output files
OUTPUTDIR="."
# You can restrict the structures that AlphaFold selects as templates
# before the release date specified as DATE
DATE="2099-07-14"
# 以降AlphaFold処理。以下は特に変更する必要なし。
mkdir -p ${OUTPUTDIR}
# ケース1:単量体予測の場合はこちらを使う
# --random_seedの数値を変えると出てくる複合体が少し変わる
alphafold --fasta_paths=${FASTAFILE} \
--output_dir=${OUTPUTDIR} \
--model_preset=monomer_ptm \
--models_to_relax=best \
--use_gpu_relax \
--random_seed=1 \
--max_template_date=${DATE}
# ケース2:複合体予測の場合はこちらを使う
# --random_seedの数値を変えると出てくる複合体が少し変わる。
# --num_multimer_predictions_per_modelの数値を増やすと
# その分予測結果が5セットずつ増える
alphafold --fasta_paths=${FASTAFILE} \
--output_dir=${OUTPUTDIR} \
--model_preset=multimer \
--models_to_relax=best \
--use_gpu_relax \
--num_multimer_predictions_per_model=1 \
--random_seed=1 \
--max_template_date=${DATE}
# ディスク容量節約のため、計算終了後のresult_model_*_pklを削除する処理(※任意)
FILE=${FASTAFILE%.*}
rm ${OUTPUTDIR}/${FILE}/result_model_*.pkl
のように設定すればOKです。
実行
構造予測したいアミノ酸配列が含まれたFASTAフォーマットのテキストファイルを用意します。例えばここでは遺伝子工学でよく使われる(使われていた?)Type-2 制限酵素 XhoIの構造予測をしてみたいと考えます。この酵素はまだ構造が明らかになっていません。
Uniprotでは https://www.uniprot.org/uniprot/Q9KVZ7 に情報がまとめられており、FASTAファイルは https://www.uniprot.org/uniprot/Q9KVZ7.fasta からダウンロードできます。
>sp|Q9KVZ7|T2X1_XANVA Type-2 restriction enzyme XhoI OS=Xanthomonas vasicola OX=56459 PE=3 SV=1
MALDLAEYDRLARLGVAQFWDGRSSALENDEERSQGGERSGVLGGRNMDGFLAMIEGIVR
KNGLPDAEVCIKGRPNLTLPGYYRPTKLWDVLVFDGKKLVAAVELKSHVGPSFGNNFNNR
AEEAIGTAHDLATAIREGILGDQLPPFTGWLILVEDCEKSKRAVRDSSPHFPVFPDFKGA
SYLTRYEVLCRKLILKGFTPRPQSLLRPALRVLGATIASFRKPRVCAHLRHGWPAMYPVG
QSRIRVNF
FASTAファイルフォーマットとは、一行目に>で始まるコメント行(何を書いても良い)、二行目以降がすべてアミノ酸配列(改行可能)となっているファイル形式です。このルールさえ守っていれば自前でFASTAファイルを書いてもOKです。これをQ9KVZ7.fastaとしてホームディレクトリ上に保存し(どこでも良い)、これについて構造予測してみます。
Alphafoldの計算時間は最低1〜3時間以上かかると思いますが、nohupコマンドをつけるとリモート先でログアウトした後でも計算し続けてくれるので便利です。alphafoldのディレクトリ上にcdで移動して、例えばホームディレクトリ上に置いてあるQ9KVZ7.fastaについて構造予測したい場合はこんな感じ。
$ nohup python3.9 docker/run_docker.py \
--fasta_paths=${HOME}/Q9KVZ7.fasta \
--max_template_date=2099-07-14 \
--model_preset=monomer_ptm \
--db_preset=full_dbs > nohup1.out &
計算結果は、Dockerfileのファイルの中の変数output_dirで指定したディレクトリに出力されます。標準出力&エラーメッセージはnohup1.outファイルに書き込まれます(この名前は自由に指定可)。
--max_template_dateの引数は必須で、指定した日までにPDBで公開された構造をAlphaFoldの構造予測のテンプレート(初期構造)として利用してくれます。この機能により、正解構造がPDBにある状態でもそれ以外の構造を元にテンプレートを選択してくれるようです。(それとは別にDeep Learningで用いる学習モデル自体は2020年5月14日までの全PDBデータを何らかの形で利用して学習しているみたいなのですが)。
--db_preset=で指定するオプションはfull_dbsかreduced_dbsの二択で、通常はfull_dbsを使うと良いでしょう。reduced_dbsはsmall_bfdを使うお試し版です。
--model_preset=で指定するオプションはAlphaFold2.1から導入されました。デフォルトはmonomerの単量体構造予測ですが、多量体を構造予測したい場合はmultimerとします。他にもmonomer用にmonomer_casp14, monomer_ptmが設定できます。
名古屋大学・不老 Type II上で動かす場合
例えば以下のようなシェルスクリプトrun_alphafold.shを書いて、任意の予測したい配列foo.fastaと同じディレクトリに置いて実行すればOKです。
参考:https://icts.nagoya-u.ac.jp/ja/sc/news/maintenance/2022-01-28-alphafold.html
ここによれば、alphafold_nvmeshというコマンドを使ったほうがalphafoldコマンドよりも高速に計算できるように設定されています。
※2023年4月5日:alphafold実行コマンドにおいて、構造最適化(relax)の引数指定が--run_relaxからver.2.3.2以降で--models_to_relax=all/best/noneに変更されています。allはすべての構造について構造最適化を実行、bestは最も予測精度が高いと考えられるモデルのみ構造最適化、noneは行わない、を表します。
#!/bin/sh
#PJM -L rscunit=cx
#PJM -L rscgrp=cxgfs-share
#PJM -L elapse=24:00:00
#PJM -j
#############################################
# ここに計算したい配列のファイル名を入力する。
FASTAFILE="foo.fasta"
# 出力先のディレクトリ名を指定する。
# '.'ならば現ディレクトリに作成する。
OUTPUTDIR="."
# テンプレート構造として使用可能なPDB構造の日付を指定。
# この日付より前に発表された構造のみをテンプレートとして使用する。
DATE="2099-07-14"
#############################################
. /usr/share/Modules/init/sh
module load cuda/11.2.1
export PATH=/center/local/app/x86/AlphaFold/v2.3.2/alphafold/bin:${PATH}
test $PJM_O_WORKDIR && cd $PJM_O_WORKDIR
mkdir -p ${OUTPUTDIR}
### 単量体予測の場合
alphafold_nvmesh \
--fasta_paths=${FASTAFILE} \
--output_dir=${OUTPUTDIR} \
--model_preset=monomer_ptm \
--models_to_relax=best \
--use_gpu_relax \
--max_template_date=${DATE}
### 複合体予測の場合
# alphafold_nvmesh \
# --fasta_paths=${FASTAFILE} \
# --output_dir=${OUTPUTDIR} \
# --model_preset=multimer \
# --models_to_relax=best \
# --use_gpu_relax \
# --num_multimer_predictions_per_model=1 \
# --max_template_date=${DATE}
###
# ディスク容量節約のため、result_model_*_pklは削除する(任意)
FILE=${FASTAFILE%.*}
rm ${OUTPUTDIR}/${FILE}/result_model_*.pkl
AlphaFold2本体のアップデート
時々AlphaFold2のプログラム自体がアップデートされることがあります。学習済みのパラメータが変更されたり、引数が変わったり、仕様が変わって便利になったりするので、基本的には最新版を使うと良いでしょう。一方で、あるバージョンをインストールし終わったあとで最新版にアップデートしたいという場合(例えばv2.3.1からv2.3.2へのアップデート)、1からAlphaFold2本体をすべて入れ直すのは苦行ですので、最低限の処理だけで済ませたいところです。
基本的には/home/moriwaki/apps/alphafold上でgit pullを行うなどして最新版のコードを反映させればアップデートできます(※dockerを使わない場合準拠)。アミノ酸配列・PDBのデータベース系は独立している外部ファイル群なので、AlphaFold2本体のコードのアップデートとは無関係です。
もしv2.3.1とv2.3.2などをバージョン分けして残しておきたい場合は以下の作業が必要になります。
- AlphaFold v2.3.1ディレクトリをコピーしてv2.3.2とする。
cp /path/to/alphafold/v2.3.1 /path/to/alphafold/v2.3.2(※v2.3.1, v.2.3.2の名前は便宜上つけた名前である) -
cd /path/to/alphafold/v2.3.2でAlphaFold v2.3.2ディレクトリに入った後、git pullなどでコードを2.3.2にアップデートする。 -
/path/to/alphafold/v2.3.2/conda/etc/profile.d/conda.shを編集して1行目export CONDA_EXE=と4行目export CONDA_PYTHON_EXEのディレクトリ名を、新しいコピー先の名前に対応するよう変更する -
/path/to/alphafold/v2.3.2/conda/bin/condaを1行目のshebangを編集して#!/path/to/alphafold/v2.3.2/conda/bin/pythonに変更する -
. conda/etc/profile.d/conda.shで更新したconda環境を読み込む -
conda activate /path/to/alphafold/v2.3.2/alphafold-condaでalphafoldのconda環境を起動 -
conda install --force-reinstall --no-deps openmm==7.7.0とし、openmmパッケージのみを再インストールする(※この再インストール作業をしないとrelaxがエラーを起こす) -
(cd ${ALPHAFOLDDIR}/alphafold-conda/lib/python3.9/site-packages/ && patch -p0 < ${ALPHAFOLDDIR}/docker/openmm.patch)を行い、OpenMM 7.5.1へのパッチを当てて構造最適化(relax)が正しく動作するようにする。
やや変則的ですが、これでアップデートできます。
結果
output_dirで指定したディレクトリに作られたrelaxed_model_*.pdbが予測構造結果です。驚きの予測精度を自身の目で確認してみましょう。
残基数380くらいのタンパク質で計算したところ計算時間129分となり(--db_preset=full_dbsの場合)、このうちhhblitsによるMSA取得が116分、最後のAlphaFold2本体による構造推論部分が13分でしたので、"GPUなしの計算機"でも気長に待てば現実的な範囲内で終わりそうです。ただいずれにしろそこそこ新しいCPUと最低32GBのRAMはほしいところです。
AlphaFoldと同日にScience誌で発表されたRoseTTAFoldとの性能比較を、PDB: 5GUEで行ってみました。AlphaFold (1枚目) とRoseTTAFold (2枚め)です。両者とも正解構造は知らない状態で予測しました(はず)。緑が予想構造で青が正解構造です。
RoseTTAFoldの正解構造からのずれを表すRMSD値は1.474Åでしたが、AlphaFold2は0.362Åでした。圧倒的精度!
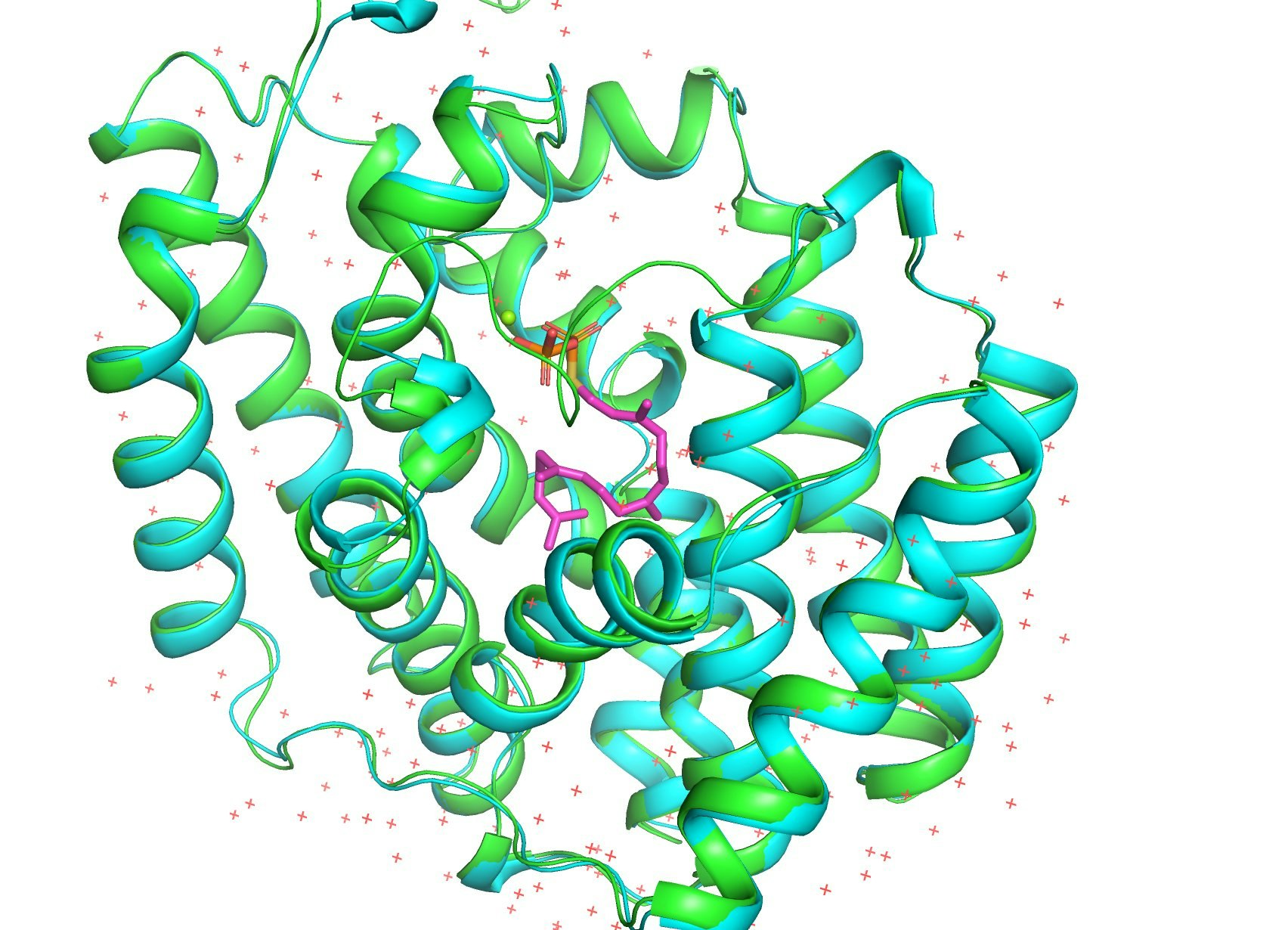
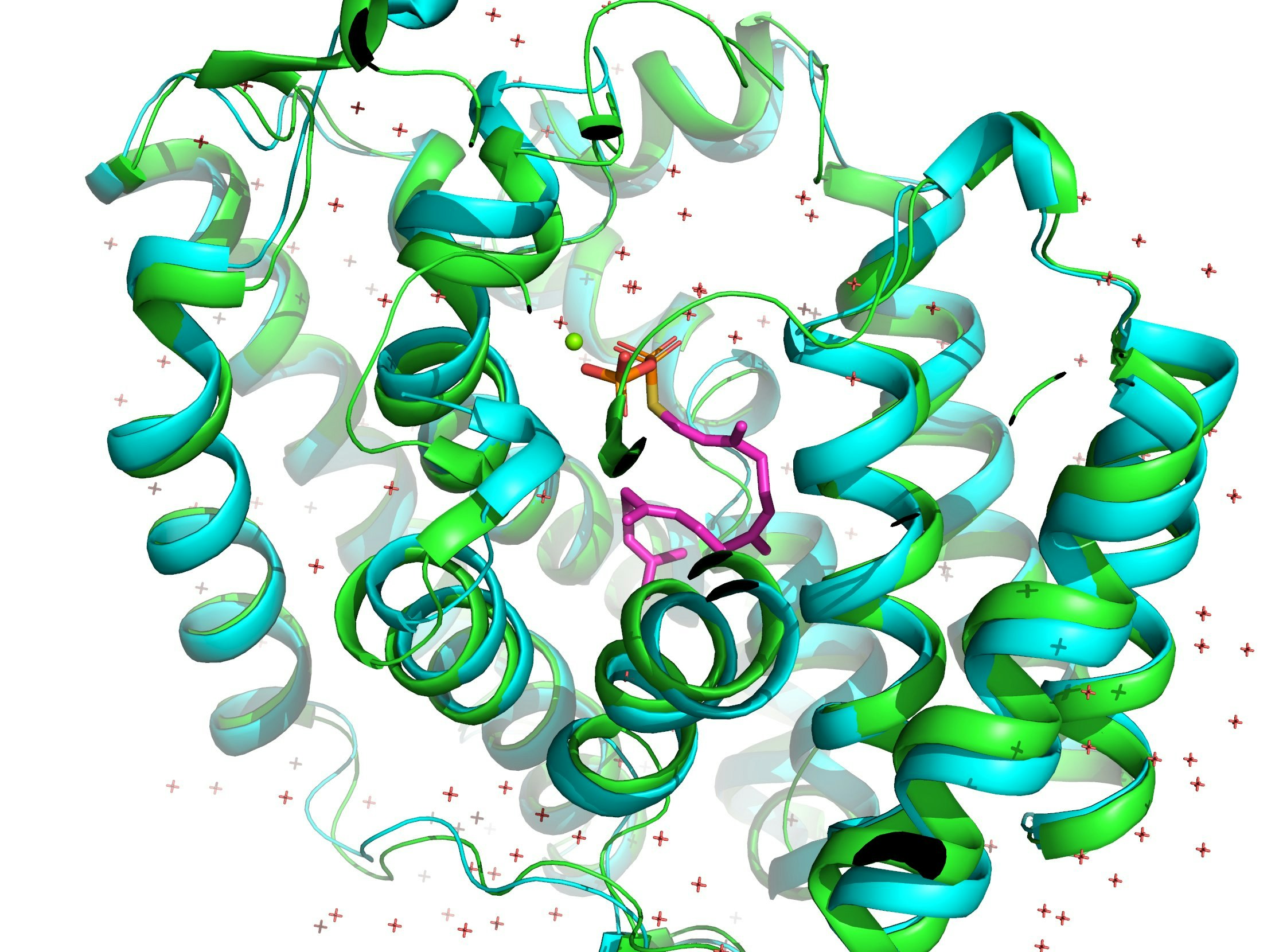
参考資料
- 英語の解説動画 Martin Steinegger氏はAlphaFold論文の著者の1人